3 R Function Mannual
3.1 Get Started
R Installation
R is a language and environment for statistical computing and graphics. We assume R (version 4.0.0 or higher) has been installed in your local machine. The latest version can be installed following instructions below for different platforms (Windows, Mac, and Linux).
-
Quick link for Windows: Download R for Windows.
-
Quick link for Mac: Download R for Mac OS X 11.
-
Below are shell command lines in Terminal (for Linux):
MNet Installation
if(!require(BiocManager)){install.packages("BiocManager")}
if (!require(remotes)) {BiocManager::install("remotes", dependencies=T)}
if (!require(devtools)) {BiocManager::install("devtools", dependencies=T)}
BiocManager::install("hfang-bristol/dnet", dependencies=T)
BiocManager::install("tuantuangui/MNet", dependencies=T)
## Check the package ‘MNet’ successfully installed
library(help=MNet)3.2 Figure 2
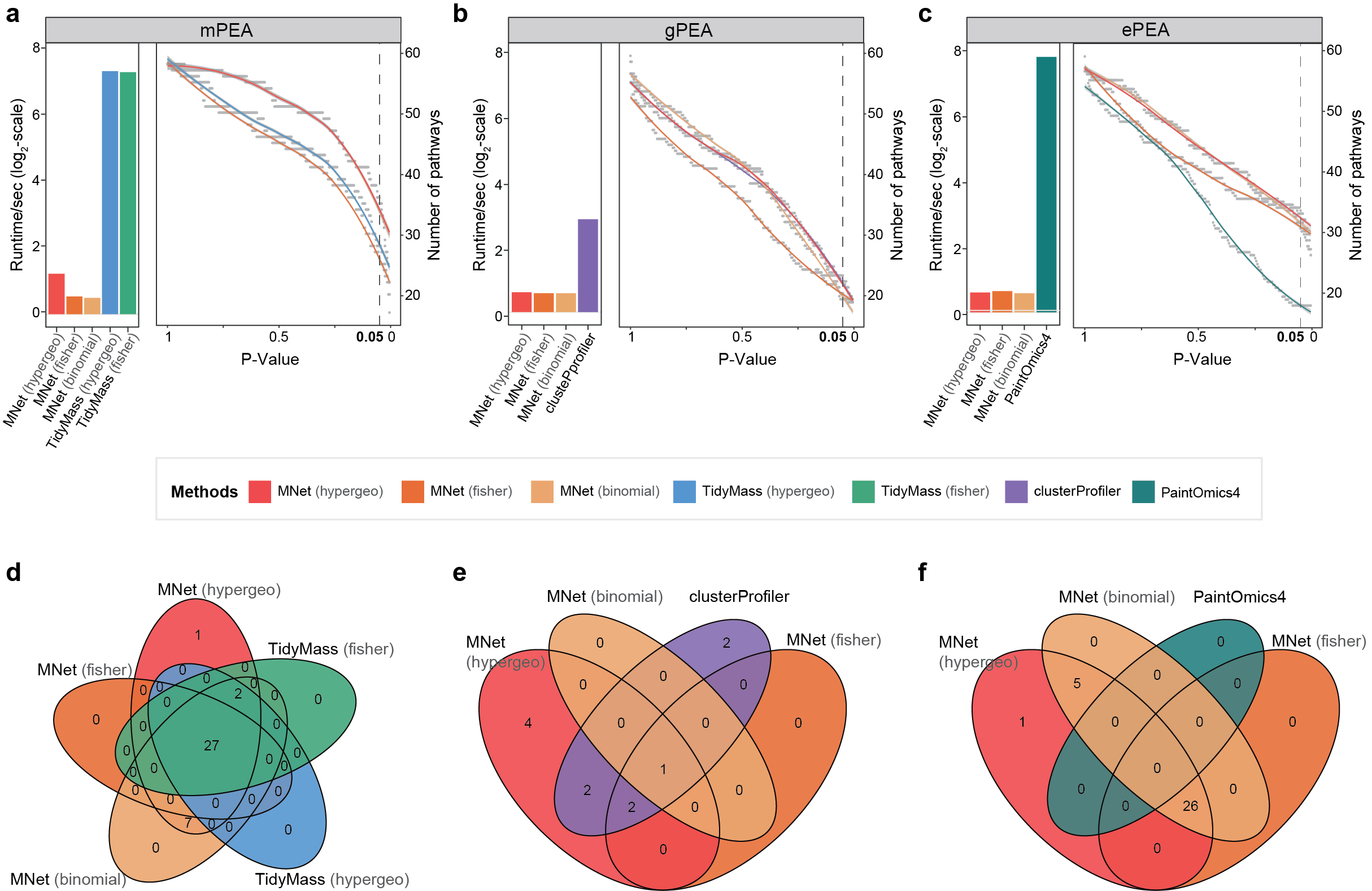
Fig. 2 | Benchmarking pathway enrichment performance.
a-c. Comparison of enriched pathway counts (line plots) and computational efficiency (log₂-scale runtime bars) between MNet and: (a) TidyMass (metabolite-based), (b) clusterProfiler (gene-based), and (c) PaintOmics (integrated).
d-f. Venn diagrams of pathway overlaps for respective comparisons.
3.2.1 (2a) TidyMass (metabolite-based)
Comparison of enriched pathway counts (line plots) and computational efficiency (log₂-scale runtime bars) between MNet and TidyMass (metabolite-based).
#-------------------------------------------------------------------------------
# Analyzing metabolites data using MNet and TidyMass and using different method parameters.
#-------------------------------------------------------------------------------
library(dplyr)
library(MNet)
all_meta <- mlimma(meta_dat,group)
## metabolite
### Filter the increase differential genes and metabolites
diff_meta <- all_meta %>%
dplyr::filter(abs(logFC) > 0.58) %>%
dplyr::filter(adj.P.Val < 0.05)
time_start <- Sys.time()
result_meta <- ePEAlyser(diff_meta$name,out="metabolite",p_cutoff=1.5,test="hypergeo")
time_end <- Sys.time()
run_time <- difftime(time_end, time_start, units = "secs")
print(paste("hypergeo_code execution time:", round(run_time, 2), "second"))
write.table(result_meta$out,"result/figure2/2a.MNet_pathway-hypergeo.txt",quote=F,row.names = F,sep="\t")
time_start <- Sys.time()
result_meta <- ePEAlyser(diff_meta$name,out="metabolite",p_cutoff=1.5,test="fisher")
time_end <- Sys.time()
run_time <- difftime(time_end, time_start, units = "secs")
print(paste("fisher—_code execution time:", round(run_time, 2), "second"))
write.table(result_meta$out,"result/figure2/2a.MNet_pathway-fisher.txt",quote=F,row.names = F,sep="\t")
time_start <- Sys.time()
result_meta <- ePEAlyser(diff_meta$name,out="metabolite",p_cutoff=1.5,test="binomial")
time_end <- Sys.time()
run_time <- difftime(time_end, time_start, units = "secs")
print(paste("binomial_code execution time:", round(run_time, 2), "second"))
write.table(result_meta$out,"result/figure2/2a.MNet_pathway-binomial.txt",quote=F,row.names = F,sep="\t")
library(tidymass)
data("kegg_hsa_pathway", package = "metpath")
pathway_database = kegg_hsa_pathway
time_start <- Sys.time()
result <- enrich_kegg(query_id = diff_meta$name,
query_type = "compound",
id_type = "KEGG",method = "hypergeometric",
pathway_database = pathway_database,
p_cutoff = 1.1,
p_adjust_method = "BH",
threads = 10)
time_end <- Sys.time()
run_time <- difftime(time_end, time_start, units = "secs")
print(paste("Tidymass-hypergeometric_code execution time:", round(run_time, 2), "second"))
write.table(result@result,"result/figure2/2a.TidyMass_pathway-hypergeometric.txt",quote=F,row.names = F,sep="\t")
time_start <- Sys.time()
result <- enrich_kegg(query_id = diff_meta$name,
query_type = "compound",
id_type = "KEGG",method = "fisher",
pathway_database = pathway_database,
p_cutoff = 1.1,
p_adjust_method = "BH",
threads = 10)
time_end <- Sys.time()
run_time <- difftime(time_end, time_start, units = "secs")
print(paste("Tidymass-fisher_code execution time:", round(run_time, 2), "second"))
write.table(result@result,"result/figure2/2a.TidyMass_pathway-fisher.txt",quote=F,row.names = F,sep="\t")
#-------------------------------------------------------------------------------
# Compare the stability of the results of different methods under different cutoff values.
#-------------------------------------------------------------------------------
library(ggplot2)
pathway_mnet_hypergeo <- data.table::fread("result/figure2/2a.MNet_pathway-hypergeo.txt") %>%
as.data.frame()
pathway_mnet_fisher <- data.table::fread("result/figure2/2a.MNet_pathway-fisher.txt") %>%
as.data.frame()
pathway_mnet_binomial <- data.table::fread("result/figure2/2a.MNet_pathway-binomial.txt") %>%
as.data.frame()
pathway_tidymass_hypergeometric <- data.table::fread("result/figure2/2a.TidyMass_pathway-hypergeometric.txt") %>%
as.data.frame() %>%
filter(pathway_name %in% PathwayExtendData$kegg_pathwayname) %>%
filter(mapped_number != 0)
pathway_tidymass_fisher <- data.table::fread("result/figure2/2a.TidyMass_pathway-fisher.txt") %>%
as.data.frame() %>%
filter(pathway_name %in% PathwayExtendData$kegg_pathwayname) %>%
filter(mapped_number != 0)
n = seq(1,0.0001,-0.005)
result_mnet_hypergeo <- data.frame()
result_mnet_fisher <- data.frame()
result_mnet_binomial <- data.frame()
result_tidymass_hypergeometric <- data.frame()
result_tidymass_fisher <- data.frame()
for (i in n) {
## hypergeo
pathway_mnet_filter <- pathway_mnet_hypergeo %>%
filter(pvalue < i)
result_mnet_temp <- data.frame(n=nrow(pathway_mnet_filter),cutoff=i)
result_mnet_hypergeo <- rbind(result_mnet_hypergeo,result_mnet_temp)
## fisher
pathway_mnet_filter <- pathway_mnet_fisher %>%
filter(pvalue < i)
result_mnet_temp <- data.frame(n=nrow(pathway_mnet_filter),cutoff=i)
result_mnet_fisher <- rbind(result_mnet_fisher,result_mnet_temp)
## binomial
pathway_mnet_filter <- pathway_mnet_binomial %>%
filter(pvalue < i)
result_mnet_temp <- data.frame(n=nrow(pathway_mnet_filter),cutoff=i)
result_mnet_binomial <- rbind(result_mnet_binomial,result_mnet_temp)
## hypergeometric
pathway_tidymass_filter <- pathway_tidymass_hypergeometric %>%
filter(p_value < i)
result_tidymass_temp <- data.frame(n=nrow(pathway_tidymass_filter),cutoff=i)
result_tidymass_hypergeometric <- rbind(result_tidymass_hypergeometric,result_tidymass_temp)
## fisher
pathway_tidymass_filter <- pathway_tidymass_fisher %>%
filter(p_value < i)
result_tidymass_temp <- data.frame(n=nrow(pathway_tidymass_filter),cutoff=i)
result_tidymass_fisher <- rbind(result_tidymass_fisher,result_tidymass_temp)
}
result <- rbind(result_mnet_hypergeo %>% mutate(type="MNet_hypergeo"),
result_mnet_fisher %>% mutate(type="MNet_fisher"),
result_mnet_binomial %>% mutate(type="MNet_binomial"),
result_tidymass_hypergeometric %>% mutate(type="tidymass_hypergeometric"),
result_tidymass_fisher %>% mutate(type="tidymass_fisher"))
p <- ggplot(result,aes(cutoff,n))+
geom_point(color="gray",size=.5)+
geom_smooth(aes(color=type))+
geom_vline(xintercept=c(0.01), linetype = 'dashed',color="gray")+
scale_color_manual(values=c("#F3B169","#f16c23","#F94141","#37AB78","#589FF3"))+
scale_x_reverse()+
theme_bw()
ggsave("result/figure2/2a.Tidymass-cutoff.pdf",p,width=6.5,height = 5)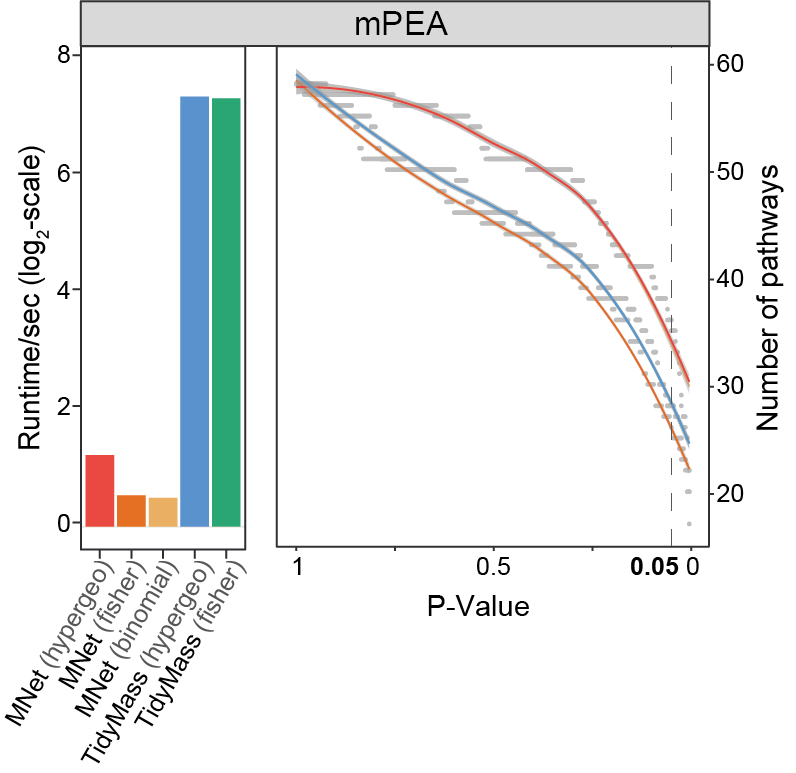
3.2.2 (2b) clusterProfiler (gene-based)
Comparison of enriched pathway counts (line plots) and computational efficiency (log₂-scale runtime bars) between MNet and clusterProfiler (gene-based).
#-------------------------------------------------------------------------------
# Analyzing metabolite-related genes data using MNet and clusterProfiler and using different method parameters.
#-------------------------------------------------------------------------------
library(clusterProfiler)
library(dplyr)
library(MNet)
library(KEGG.db)
detach(package:xcms)
all_gene <- mlimma(gene_dat,group)
gene_filter <- PathwayExtendData %>%
filter(type=="gene") %>%
pull(name) %>% unique()
## gene
### Filter the increase differential genes and metabolites
diff_gene_all <- all_gene %>%
dplyr::filter(abs(logFC) > 0.58) %>%
dplyr::filter(adj.P.Val < 0.05)
gene.data <- clusterProfiler::bitr(diff_gene_all$name,fromType = "SYMBOL",toType ="ENTREZID",OrgDb='org.Hs.eg.db')
diff_gene_clusterprofiler <- diff_gene_all %>%
inner_join(gene.data,by=c("name"="SYMBOL"))
kegg_F <- enrichKEGG(
gene = diff_gene_clusterprofiler$ENTREZID,
keyType = 'kegg',
organism = 'hsa',
minGSSize = 1,
pAdjustMethod = 'BH',
pvalueCutoff = 1.1,
use_internal_data = F)
write.table(kegg_F@result,"result/figure2/2b.clusterprofiler-gene-F.txt",quote=F,row.names = F,sep="\t")
kegg_T <- enrichKEGG(
gene = diff_gene_clusterprofiler$ENTREZID,
keyType = 'kegg',
organism = 'hsa',
minGSSize = 1,
pAdjustMethod = 'BH',
pvalueCutoff = 1.1,
use_internal_data = T)
write.table(kegg_T@result,"result/figure2/2b.clusterprofiler-gene-T.txt",quote=F,row.names = F,sep="\t")
diff_gene_mnet <- diff_gene_all %>%
filter(name %in% gene_filter)
gene_MNet_result_hypergeo <- ePEAlyser(diff_gene_mnet$name,out="gene",p_cutoff=1.1,test="hypergeo")
write.table(gene_MNet_result_hypergeo$output,"result/figure2/2b.MNet-gene_hypergeo.txt",quote=F,row.names = F,sep="\t")
gene_MNet_result_fisher <- ePEAlyser(diff_gene_mnet$name,out="gene",p_cutoff=1.1,test="fisher")
write.table(gene_MNet_result_fisher$output,"result/figure2/2b.MNet-gene_fisher.txt",quote=F,row.names = F,sep="\t")
gene_MNet_result_binomial <- ePEAlyser(diff_gene_mnet$name,out="gene",p_cutoff=1.1,test="binomial")
write.table(gene_MNet_result_binomial$output,"result/figure2/2b.MNet-gene_binomial.txt",quote=F,row.names = F,sep="\t")
#-------------------------------------------------------------------------------
# Compare the stability of the results of different methods under different cutoff values.
#-------------------------------------------------------------------------------
library(ggplot2)
pathway_mnet_hypergeo <- data.table::fread("result/figure2/2b.MNet-gene_hypergeo.txt") %>%
as.data.frame()
pathway_mnet_fisher <- data.table::fread("result/figure2/2b.MNet-gene_fisher.txt") %>%
as.data.frame()
pathway_mnet_binomial <- data.table::fread("result/figure2/2b.MNet-gene_binomial.txt") %>%
as.data.frame()
pathway_clusterprofiler_F <- data.table::fread("result/figure2/2b.clusterprofiler-gene-F.txt") %>%
as.data.frame() %>%
filter(ID %in% PathwayExtendData$kegg_pathwayid)
pathway_clusterprofiler_T <- data.table::fread("result/figure2/2b.clusterprofiler-gene-T.txt") %>%
as.data.frame() %>%
filter(ID %in% PathwayExtendData$kegg_pathwayid)
n = seq(1,0.0001,-0.005)
result_mnet_hypergeo <- data.frame()
result_mnet_fisher <- data.frame()
result_mnet_binomial <- data.frame()
result_clusterprofiler_T <- data.frame()
result_clusterprofiler_F <- data.frame()
for (i in n) {
## hypergeo
pathway_mnet_hypergeo_filter <- pathway_mnet_hypergeo %>%
filter(pvalue < i)
result_mnet_hypergeo_temp <- data.frame(n=nrow(pathway_mnet_hypergeo_filter),cutoff=i)
result_mnet_hypergeo <- rbind(result_mnet_hypergeo,result_mnet_hypergeo_temp)
## fisher
pathway_mnet_fisher_filter <- pathway_mnet_fisher %>%
filter(pvalue < i)
result_mnet_fisher_temp <- data.frame(n=nrow(pathway_mnet_fisher_filter),cutoff=i)
result_mnet_fisher <- rbind(result_mnet_fisher,result_mnet_fisher_temp)
## binomial
pathway_mnet_binomial_filter <- pathway_mnet_binomial %>%
filter(pvalue < i)
result_mnet_binomial_temp <- data.frame(n=nrow(pathway_mnet_binomial_filter),cutoff=i)
result_mnet_binomial <- rbind(result_mnet_binomial,result_mnet_binomial_temp)
# clusterProfiler-T
pathway_clusterprofiler_T_filter <- pathway_clusterprofiler_T %>%
filter(pvalue < i)
result_clusterprofiler_T_temp <- data.frame(n=nrow(pathway_clusterprofiler_T_filter),cutoff=i)
result_clusterprofiler_T <- rbind(result_clusterprofiler_T,result_clusterprofiler_T_temp)
# clusterProfiler-F
pathway_clusterprofiler_F_filter <- pathway_clusterprofiler_F %>%
filter(pvalue < i)
result_clusterprofiler_F_temp <- data.frame(n=nrow(pathway_clusterprofiler_F_filter),cutoff=i)
result_clusterprofiler_F <- rbind(result_clusterprofiler_F,result_clusterprofiler_F_temp)
}
result <- rbind(result_mnet_hypergeo %>% mutate(type="MNet_hypergeo"),
result_mnet_fisher %>% mutate(type="MNet_fisher"),
result_mnet_binomial %>% mutate(type="MNet_binomial"),
result_clusterprofiler_F %>% mutate(type="clusterprofiler_F"))
p <- ggplot(result,aes(cutoff,n))+
geom_point(color="gray",size=.5)+
geom_smooth(aes(color=type))+
geom_vline(xintercept=c(0.01), linetype = 'dashed',color="gray")+
scale_color_manual(values=c("MNet_hypergeo"="#F94141",
"MNet_fisher"="#f16c23",
"MNet_binomial"="#F3B169",
"clusterprofiler_F"="#AA66EB"))+
scale_x_reverse()+
theme_bw()
ggsave("result/figure2/2b.clusterprofiler-cutoff.pdf",p,width=6,height = 5)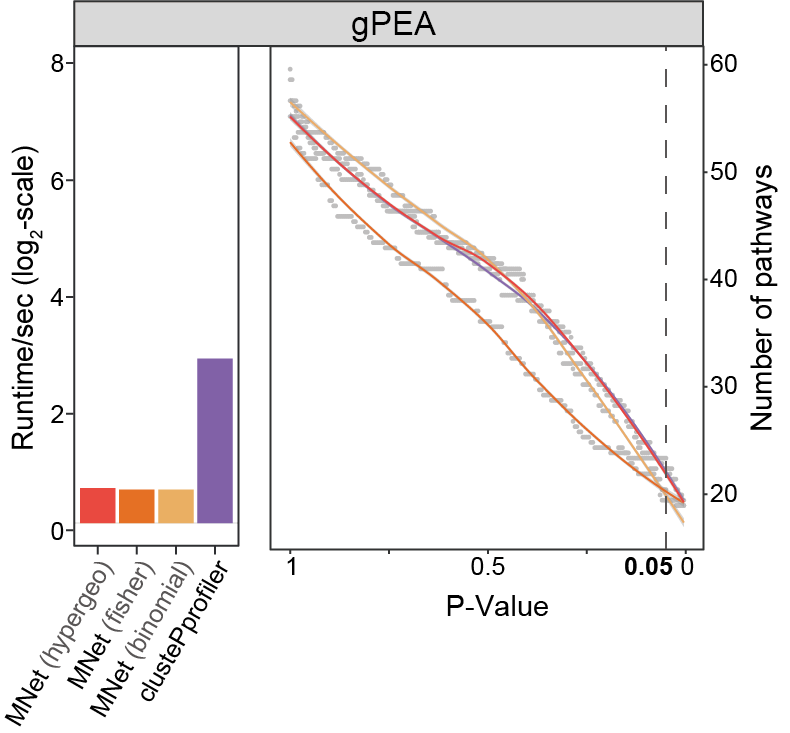
3.2.3 (2c) PaintOmics (integrated)
Comparison of enriched pathway counts (line plots) and computational efficiency (log₂-scale runtime bars) between MNet and PaintOmics (integrated).
#-------------------------------------------------------------------------------
# Analyzing metabolites and metabolite-related genes data using MNet and PaintOmics and using different method parameters.
#-------------------------------------------------------------------------------
library(dplyr)
library(MNet)
all_meta <- mlimma(meta_dat,group)
## metabolite
### Filter the increase differential genes and metabolites
diff_meta <- all_meta %>%
dplyr::filter(abs(logFC) > 0.58) %>%
dplyr::filter(adj.P.Val < 0.05)
all_gene <- mlimma(gene_dat,group)
## gene
## Filter the increase differential genes and metabolites
diff_gene <- all_gene %>%
dplyr::filter(abs(logFC) > 0.58) %>%
dplyr::filter(adj.P.Val < 0.05)
result <- rbind(diff_meta,diff_gene) %>%
filter(name %in% PathwayExtendData$name)
result_pathway_hypergeo <- ePEAlyser(result$name,out="Extended",p_cutoff=1.1,test="hypergeo")
write.table(result_pathway_hypergeo$output,"result/figure2/2c.MNet-Extended-hypergeo.txt",quote=F,row.names = F,sep="\t")
result_pathway_fisher <- ePEAlyser(result$name,out="Extended",p_cutoff=1.1,test="fisher")
write.table(result_pathway_fisher$output,"result/figure2/2c.MNet-Extended-fisher.txt",quote=F,row.names = F,sep="\t")
result_pathway_binomial <- ePEAlyser(result$name,out="Extended",p_cutoff=1.1,test="binomial")
write.table(result_pathway_binomial$output,"result/figure2/2c.MNet-Extended-binomial.txt",quote=F,row.names = F,sep="\t")
#-------------------------------------------------------------------------------
# Compare the stability of the results of different methods under different cutoff values.
#-------------------------------------------------------------------------------
library(ggplot2)
pathway_mnet_hypergeo <- data.table::fread("result/figure2/2c.MNet-Extended-hypergeo.txt") %>%
as.data.frame()
pathway_mnet_fisher <- data.table::fread("result/figure2/2c.MNet-Extended-fisher.txt") %>%
as.data.frame()
pathway_mnet_binomial <- data.table::fread("result/figure2/2c.MNet-Extended-binomial.txt") %>%
as.data.frame()
pathway_paintomics <- data.table::fread("result/figure2/2c.PaintOmics.txt") %>%
as.data.frame() %>%
dplyr::select(-V7) %>%
dplyr::filter(`Pathway name` %in% PathwayExtendData$kegg_pathwayname)
n = seq(1,0.0001,-0.005)
result_mnet_hypergeo <- data.frame()
result_mnet_fisher <- data.frame()
result_mnet_binomial <- data.frame()
result_paintomics <- data.frame()
for (i in n) {
## hypergeo
pathway_mnet_filter <- pathway_mnet_hypergeo %>%
filter(pvalue < i)
result_mnet_temp <- data.frame(n=nrow(pathway_mnet_filter),cutoff=i)
result_mnet_hypergeo <- rbind(result_mnet_hypergeo,result_mnet_temp)
## fisher
pathway_mnet_filter <- pathway_mnet_fisher %>%
filter(pvalue < i)
result_mnet_temp <- data.frame(n=nrow(pathway_mnet_filter),cutoff=i)
result_mnet_fisher <- rbind(result_mnet_fisher,result_mnet_temp)
## binomial
pathway_mnet_filter <- pathway_mnet_binomial %>%
filter(pvalue < i)
result_mnet_temp <- data.frame(n=nrow(pathway_mnet_filter),cutoff=i)
result_mnet_binomial <- rbind(result_mnet_binomial,result_mnet_temp)
## hypergeometric
pathway_paintomics_filter <- pathway_paintomics %>%
filter(`Combined pValue(Fisher)` < i)
result_paintomics_temp <- data.frame(n=nrow(pathway_paintomics_filter),cutoff=i)
result_paintomics <- rbind(result_paintomics,result_paintomics_temp)
}
result <- rbind(result_mnet_hypergeo %>% mutate(type="MNet_hypergeo"),
result_mnet_fisher %>% mutate(type="MNet_fisher"),
result_mnet_binomial %>% mutate(type="MNet_binomial"),
result_paintomics %>% mutate(type="paintomics"))
p <- ggplot(result,aes(cutoff,n))+
geom_point(color="gray",size=.5)+
geom_smooth(aes(color=type))+
geom_vline(xintercept=c(0.01), linetype = 'dashed',color="gray")+
scale_color_manual(values=c("#F3B169","#f16c23","#F94141","#008080"))+
scale_x_reverse()+
theme_bw()
ggsave("result/figure2/2c.PaintOmics-cutoff.pdf",p,width=6.5,height = 5)
3.2.4 (2d-f) Venn diagrams
Venn diagrams of pathway overlaps for respective comparisons.
library(VennDiagram)
#-------------------------------------------------------------------------------
# TidyMass
#-------------------------------------------------------------------------------
pathway_mnet_hypergeo <- data.table::fread("result/figure2/2a.MNet_pathway-hypergeo.txt") %>%
as.data.frame() %>%
filter(pvalue < 0.05)
pathway_mnet_fisher <- data.table::fread("result/figure2/2a.MNet_pathway-fisher.txt") %>%
as.data.frame() %>%
filter(pvalue < 0.05)
pathway_mnet_binomial <- data.table::fread("result/figure2/2a.MNet_pathway-binomial.txt") %>%
as.data.frame() %>%
filter(pvalue < 0.05)
pathway_tidymass_hypergeometric <- data.table::fread("result/figure2/2a.TidyMass_pathway-hypergeometric.txt") %>%
as.data.frame() %>%
filter(pathway_name %in% PathwayExtendData$kegg_pathwayname) %>%
dplyr::filter(p_value < 0.05)
pathway_tidymass_fisher <- data.table::fread("result/figure2/2a.TidyMass_pathway-fisher.txt") %>%
as.data.frame() %>%
filter(pathway_name %in% PathwayExtendData$kegg_pathwayname) %>%
dplyr::filter(p_value < 0.05)
venn.plot <- venn.diagram(
x = list(
MNet_hypergeo = pathway_mnet_hypergeo$name,
MNet_fisher = pathway_mnet_fisher$name,
MNet_binomial = pathway_mnet_binomial$name,
tidymass_hypergeometric = pathway_tidymass_hypergeometric$pathway_name,
tidymass_fisher = pathway_tidymass_fisher$pathway_name),
filename = NULL,
fill = c("#F94141","#f16c23","#F3B169","#589FF3","#37AB78"),
alpha = 0.9)
pdf("result/figure2/2d.MNet-TidyMass-venn.pdf",width=7,height = 7)
grid.draw(venn.plot)
dev.off()
#-------------------------------------------------------------------------------
# clusterProfiler
#-------------------------------------------------------------------------------
pathway_mnet_hypergeo <- data.table::fread("result/figure2/2b.MNet-gene_hypergeo.txt") %>%
as.data.frame() %>%
dplyr::select(name,pvalue) %>%
mutate(type="MNet_hypergeo") %>%
filter(pvalue < 0.05)
pathway_mnet_fisher <- data.table::fread("result/figure2/2b.MNet-gene_fisher.txt") %>%
as.data.frame() %>%
dplyr::select(name,pvalue) %>%
mutate(type="MNet_fisher") %>%
filter(pvalue < 0.05)
pathway_mnet_binomial <- data.table::fread("result/figure2/2b.MNet-gene_binomial.txt") %>%
as.data.frame() %>%
dplyr::select(name,pvalue) %>%
mutate(type="MNet_binomial") %>%
filter(pvalue < 0.05)
pathway_clusterprofiler <- data.table::fread("result/figure2/2b.clusterprofiler-gene-F.txt") %>%
as.data.frame() %>%
filter(ID %in% PathwayExtendData$kegg_pathwayid) %>%
dplyr::select(Description,pvalue) %>%
rename("name"="Description") %>%
mutate(type="clusterprofiler") %>%
filter(pvalue < 0.05)
a<-venn.diagram(list(mnet_hypergeo=pathway_mnet_hypergeo$name,
mnet_fisher=pathway_mnet_fisher$name,
mnet_binomial=pathway_mnet_binomial$name,
clusterprofiler=pathway_clusterprofiler$name),
filename=NULL,fill=c("#F94141","#f16c23","#F3B169","#AA66EB"),alpha =0.9)
pdf("result/figure2/2f.clusterprofiler-venn.pdf",width=5,height = 5)
grid.draw(a)
dev.off()
#-------------------------------------------------------------------------------
# PaintOmics
#-------------------------------------------------------------------------------
pathway_mnet_hypergeo <- data.table::fread("result/figure2/2c.MNet-Extended-hypergeo.txt") %>%
as.data.frame() %>%
filter(pvalue < 0.05)
pathway_mnet_fisher <- data.table::fread("result/figure2/2c.MNet-Extended-fisher.txt") %>%
as.data.frame() %>%
filter(pvalue < 0.05)
pathway_mnet_binomial <- data.table::fread("result/figure2/2c.MNet-Extended-binomial.txt") %>%
as.data.frame() %>%
filter(pvalue < 0.05)
## Running metabolite pathway analysis at PaintOmics4 website
pathway_paintomics <- data.table::fread("result/figure2/2c.PaintOmics.txt") %>%
as.data.frame() %>%
filter(`Pathway name` %in% PathwayExtendData$kegg_pathwayname) %>%
dplyr::filter(`Combined pValue(Fisher)` < 0.05)
venn.plot <- venn.diagram(
x = list(
MNet_hypergeo = pathway_mnet_hypergeo$name,
MNet_fisher = pathway_mnet_fisher$name,
MNet_binomial = pathway_mnet_binomial$name,
PaintOmics = pathway_paintomics$`Pathway name`),
filename = NULL,
fill = c("#F94141","#f16c23","#F3B169","#008080"),
alpha = 0.9)
pdf("result/figure2/2f.MNet-PaintOmics-venn.pdf",width=7,height = 7)
grid.draw(venn.plot)
dev.off()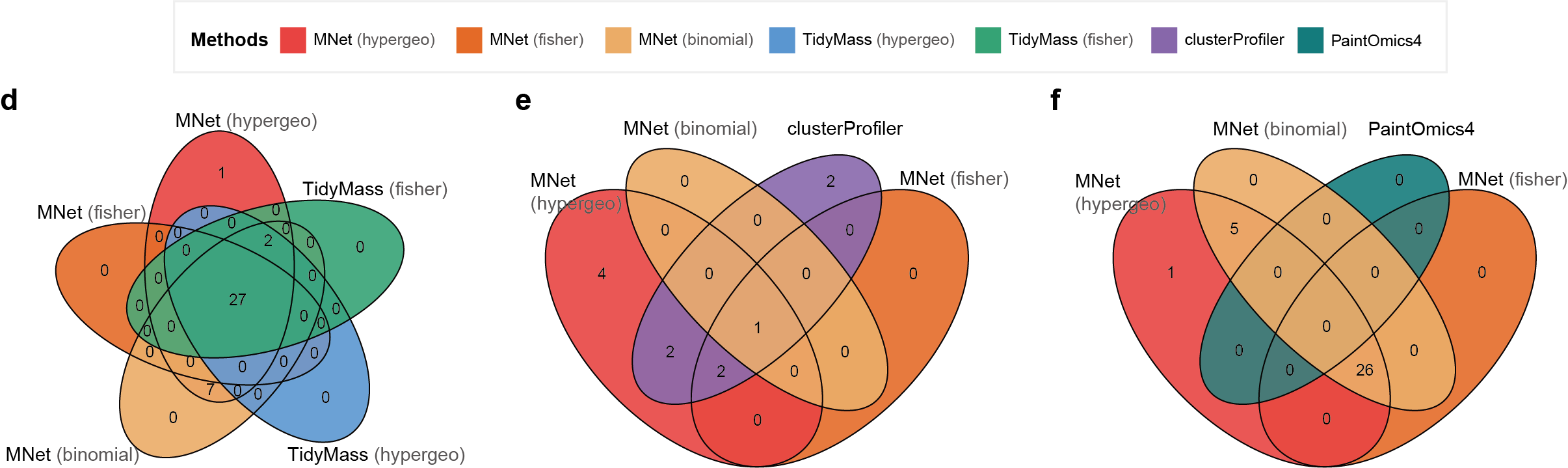
3.3 Figure 3

Fig. 3 | Integrated gene set enrichment analysis enhances pathway detection.
- Metabolic pathway counts identified by MNet (eSEA) versus multiGSEA across input types.
b-d. Pathway overlaps for: (b) metabolite-based, (c) gene-based, and (d) integrated metabolite-gene analyses.
3.3.1 (3a) Metabolic pathway counts
Metabolic pathway counts identified by MNet (eSEA) versus multiGSEA across input types.
#-------------------------------------------------------------------------------
# Analyzing metabolites and metabolite-related genes data using MNet and using different method parameters.
#-------------------------------------------------------------------------------
library(multiGSEA)
library(org.Hs.eg.db)
library(dplyr)
library(MNet)
data(transcriptome,package="multiGSEA")
data(metabolome,package="multiGSEA")
ah <- AnnotationHub::AnnotationHub()
aa <- ah[["AH91792"]]
metabolome_1 <- metabolome %>%
mutate(HMDB=gsub("HMDB","HMDB00",HMDB)) %>%
left_join(aa,by="HMDB") %>%
filter(!is.na(KEGG)) %>%
as.data.frame() %>%
dplyr::select(HMDB,logFC,pValue,KEGG,HMDB) %>%
unique() %>%
filter(KEGG %in% PathwayExtendData$name) %>%
distinct(KEGG,.keep_all = T) %>%
arrange(desc(logFC)) %>%
distinct(logFC,.keep_all = T)
dat_metabolome <- metabolome_1$logFC
names(dat_metabolome) <- metabolome_1$KEGG
set.seed(1)
result_metabolome <- eSEAlyser(dat_metabolome,minSize = 1,out="metabolite",gseaParam=1,nPermSimple=1000)
transcriptome_temp <- transcriptome %>%
as.data.frame() %>%
filter(Symbol %in% PathwayExtendData$name) %>%
arrange(desc(logFC))
dat_transcriptome <- transcriptome_temp$logFC
names(dat_transcriptome) <- transcriptome_temp$Symbol
set.seed(1)
result_transcriptome <- eSEAlyser(dat_transcriptome,minSize = 1,out="gene",gseaParam=1,nPermSimple=1000)
dat_all_temp <- rbind(metabolome_1 %>%
dplyr::select(KEGG,logFC) %>%
dplyr::rename("name"="KEGG"),
transcriptome %>%
as.data.frame() %>%
dplyr::select(Symbol,logFC) %>%
dplyr::rename("name"="Symbol")) %>%
filter(name %in% PathwayExtendData$name) %>%
arrange(desc(logFC))
dat_all <- dat_all_temp$logFC
names(dat_all) <- dat_all_temp$name
set.seed(1)
result_extended <- eSEAlyser(dat_all,minSize = 1,out="Extended",gseaParam=1,nPermSimple=1000)
write.table(result_metabolome,"result/figure3/3a.MNet_GSEA_metabolome.txt",quote=F,row.names=F,sep="\t")
write.table(result_transcriptome,"result/figure3/3a.MNet_GSEA_transcriptome.txt",quote=F,row.names=F,sep="\t")
write.table(result_extended,"result/figure3/3a.MNet_GSEA_extended.txt",quote=F,row.names=F,sep="\t")
#-------------------------------------------------------------------------------
# Analyzing metabolites and metabolite-related genes data using multiGSEA and using different method parameters.
#-------------------------------------------------------------------------------
## different
data(transcriptome,package="multiGSEA")
data(metabolome,package="multiGSEA")
head(transcriptome)
head(metabolome)
transcriptome <- transcriptome %>%
arrange(desc(logFC))
ah <- AnnotationHub::AnnotationHub()
aa <- ah[["AH91792"]]
metabolome <- metabolome %>%
mutate(HMDB=gsub("HMDB","HMDB00",HMDB)) %>%
left_join(aa,by="HMDB") %>%
filter(!is.na(KEGG)) %>%
as.data.frame() %>%
dplyr::select(HMDB,logFC,pValue,KEGG,HMDB) %>%
unique() %>%
distinct(KEGG,.keep_all = T) %>%
arrange(desc(logFC)) %>%
distinct(logFC,.keep_all = T)
## create data structure
omics_data <- initOmicsDataStructure(layer = c("transcriptome","metabolome"))
## add transcriptome layer
omics_data$transcriptome <- transcriptome$logFC
names(omics_data$transcriptome) <- transcriptome$Symbol
## add metabolome layer
## HMDB features have to be updated to the new HMDB format
omics_data$metabolome <- metabolome$logFC
names(omics_data$metabolome) <- metabolome$KEGG
databases <- c("kegg")
layers <- names(omics_data)
pathways <- getMultiOmicsFeatures(
dbs = databases, layer = layers,
returnTranscriptome = "SYMBOL",
returnMetabolome = "KEGG",
useLocal = T)
set.seed(1)
enrichment_scores <- multiGSEA(pathways, omics_data)
df <- extractPvalues(
enrichmentScores = enrichment_scores,
pathwayNames = names(pathways[[1]]))
df$combined_pval <- combinePvalues(df)
df$combined_padj <- p.adjust(df$combined_pval, method = "BH")
df <- cbind(data.frame(pathway = names(pathways[[1]])), df)
write.table(df,"result/figure3/3a.multiGSEA-diff.txt",quote=F,row.names = F,sep="\t")
## same
data(transcriptome,package="multiGSEA")
data(metabolome,package="multiGSEA")
head(transcriptome)
head(metabolome)
transcriptome <- transcriptome %>%
filter(Symbol %in% PathwayExtendData$name) %>%
arrange(desc(logFC))
ah <- AnnotationHub::AnnotationHub()
aa <- ah[["AH91792"]]
metabolome <- metabolome %>%
mutate(HMDB=gsub("HMDB","HMDB00",HMDB)) %>%
left_join(aa,by="HMDB") %>%
filter(!is.na(KEGG)) %>%
as.data.frame() %>%
dplyr::select(HMDB,logFC,pValue,KEGG,HMDB) %>%
unique() %>%
filter(KEGG %in% PathwayExtendData$name) %>%
distinct(KEGG,.keep_all = T) %>%
arrange(desc(logFC)) %>%
distinct(logFC,.keep_all = T)
# create data structure
omics_data <- initOmicsDataStructure(layer = c("transcriptome","metabolome"))
## add transcriptome layer
omics_data$transcriptome <- transcriptome$logFC
names(omics_data$transcriptome) <- transcriptome$Symbol
## add metabolome layer
## HMDB features have to be updated to the new HMDB format
omics_data$metabolome <- metabolome$logFC
names(omics_data$metabolome) <- metabolome$KEGG
pathways <- list()
pathways$transcriptome <- Pathways_gene
pathways$metabolome <- Pathways_metabolite
set.seed(1)
enrichment_scores <- multiGSEA(pathways, omics_data)
df <- extractPvalues(
enrichmentScores = enrichment_scores,
pathwayNames = names(pathways[[1]]))
df$combined_pval <- combinePvalues(df)
df$combined_padj <- p.adjust(df$combined_pval, method = "BH")
df <- cbind(data.frame(pathway = names(pathways[[1]])), df)
write.table(df,"result/figure3/3a.multiGSEA-same.txt",quote=F,row.names = F,sep="\t")
#-------------------------------------------------------------------------------
# Compare the stability of the results of different methods under different cutoff values.
#-------------------------------------------------------------------------------
library(ggplot2)
dat_mnet_gene <- data.table::fread("result/figure3/3a.MNet_GSEA_transcriptome.txt") %>%
as.data.frame() %>%
mutate(type="gene",software="MNet")
dat_mnet_metabolite <- data.table::fread("result/figure3/3a.MNet_GSEA_metabolome.txt") %>%
as.data.frame() %>%
mutate(type="metabolite",software="MNet")
dat_mnet_extended <- data.table::fread("result/figure3/3a.MNet_GSEA_extended.txt") %>%
as.data.frame() %>%
mutate(type="extended",software="MNet")
dat_mnet_all <- rbind(dat_mnet_gene,dat_mnet_metabolite,dat_mnet_extended) %>%
dplyr::select(pathway,pval,type,software)
dat_multigsea <- data.table::fread("result/figure3/3a.multiGSEA-diff.txt") %>%
as.data.frame() %>%
mutate(pathway=substr(pathway, 8, nchar(pathway))) %>%
filter(pathway %in% PathwayExtendData$kegg_pathwayname) %>%
dplyr::select(pathway,transcriptome_pval,metabolome_pval,combined_pval)
dat_multigsea_melt <- reshape2::melt(dat_multigsea,id="pathway")
dat_multigsea_same <- dat_multigsea_melt %>%
dplyr::rename("pval"="value","type"="variable") %>%
mutate(software="multiGSEA-diffdatabase") %>%
mutate(type=ifelse(type=="transcriptome_pval","gene",
ifelse(type=="metabolome_pval","metabolite",
ifelse(type=="combined_pval","extended","jj"))))
## same database
dat_multigsea <- data.table::fread("result/figure3/3a.multiGSEA-same.txt") %>%
as.data.frame() %>%
filter(pathway %in% PathwayExtendData$kegg_pathwayname) %>%
dplyr::select(pathway,transcriptome_pval,metabolome_pval,combined_pval)
dat_multigsea_melt <- reshape2::melt(dat_multigsea,id="pathway")
dat_multigsea_diff <- dat_multigsea_melt %>%
dplyr::rename("pval"="value","type"="variable") %>%
mutate(software="multiGSEA-samedatabase") %>%
mutate(type=ifelse(type=="transcriptome_pval","gene",
ifelse(type=="metabolome_pval","metabolite",
ifelse(type=="combined_pval","extended","jj"))))
all <- rbind(dat_mnet_all,dat_multigsea_same,dat_multigsea_diff)
n = seq(1,0.0001,-0.005)
result <- data.frame()
for (i in n) {
## Hypergeo
result_temp <- all %>%
filter(pval < i) %>%
group_by(type,software) %>%
summarise(n=n()) %>%
as.data.frame() %>%
mutate(cutoff=i)
result <- rbind(result,result_temp)
}
result$type <- factor(result$type,levels=c("metabolite","gene","extended"))
p <- ggplot(result,aes(cutoff,n))+
geom_point(color="gray",size=.5)+
geom_smooth(aes(color=software))+
geom_vline(xintercept=c(0.01), linetype = 'dashed',color="gray")+
facet_grid(. ~ type)+
scale_color_manual(values=c("MNet"="#EF2C2B",
"multiGSEA-diffdatabase"="#4FBD81",
"multiGSEA-samedatabase"="#FFC839"))+
scale_x_reverse()+
theme_bw()
ggsave("result/figure3/3a.MNet-multiGSEA-cutoff.pdf",p,width=12,height = 5)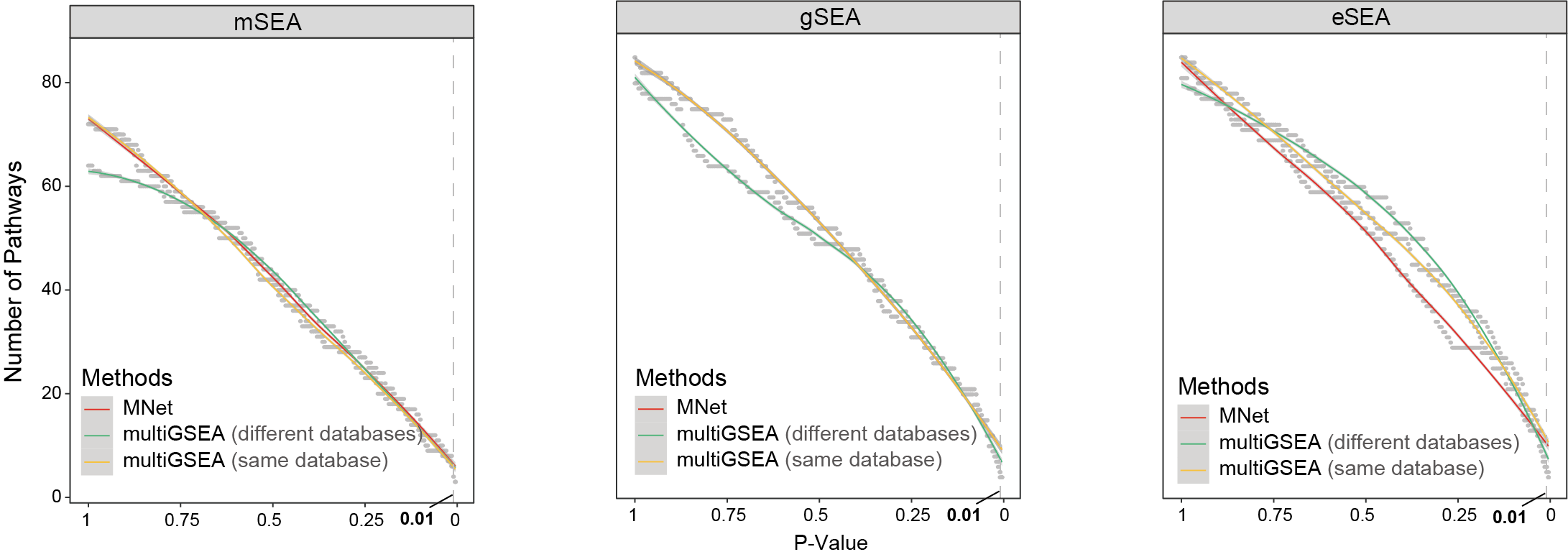
3.3.2 (3b-d) Venn diagrams
Pathway overlaps for: (b) metabolite-based, (c) gene-based, and (d) integrated metabolite-gene analyses.
library(VennDiagram)
#-------------------------------------------------------------------------------
# Load data
#-------------------------------------------------------------------------------
dat_mnet_gene <- data.table::fread("result/figure3/3a.MNet_GSEA_transcriptome.txt") %>%
as.data.frame() %>%
mutate(type="gene",software="MNet")
dat_mnet_metabolite <- data.table::fread("result/figure3/3a.MNet_GSEA_metabolome.txt") %>%
as.data.frame() %>%
mutate(type="metabolite",software="MNet")
dat_mnet_extended <- data.table::fread("result/figure3/3a.MNet_GSEA_extended.txt") %>%
as.data.frame() %>%
mutate(type="extended",software="MNet")
dat_mnet_all <- rbind(dat_mnet_gene,dat_mnet_metabolite,dat_mnet_extended) %>%
dplyr::select(pathway,pval,type,software)
dat_multigsea <- data.table::fread("result/figure3/3a.multiGSEA-diff.txt") %>%
as.data.frame() %>%
mutate(pathway=substr(pathway, 8, nchar(pathway))) %>%
filter(pathway %in% PathwayExtendData$kegg_pathwayname) %>%
dplyr::select(pathway,transcriptome_pval,metabolome_pval,combined_pval)
dat_multigsea_melt <- reshape2::melt(dat_multigsea,id="pathway")
dat_multigsea_diff <- dat_multigsea_melt %>%
dplyr::rename("pval"="value","type"="variable") %>%
dplyr::mutate(software="multiGSEA-diffdatabase") %>%
dplyr::mutate(type=ifelse(type=="transcriptome_pval","gene",
ifelse(type=="metabolome_pval","metabolite", ifelse(type=="combined_pval","extended","jj"))))
dat_multigsea <- data.table::fread("result/figure3/3a.multiGSEA-same.txt") %>%
as.data.frame() %>%
filter(pathway %in% PathwayExtendData$kegg_pathwayname) %>%
dplyr::select(pathway,transcriptome_pval,metabolome_pval,combined_pval)
dat_multigsea_melt <- reshape2::melt(dat_multigsea,id="pathway")
dat_multigsea_same <- dat_multigsea_melt %>%
dplyr::rename("pval"="value","type"="variable") %>%
dplyr::mutate(software="multiGSEA-samedatabase") %>%
dplyr::mutate(type=ifelse(type=="transcriptome_pval","gene",
ifelse(type=="metabolome_pval","metabolite", ifelse(type=="combined_pval","extended","jj"))))
all <- rbind(dat_mnet_all,dat_multigsea_diff,dat_multigsea_same)
all_filter <- all %>%
filter(pval < 0.01)
all_filter_metabolite <- all_filter %>%
filter(type=="metabolite")
all_filter_gene <- all_filter %>%
filter(type=="gene")
all_filter_extended <- all_filter %>%
filter(type=="extended")
#-------------------------------------------------------------------------------
# metabolite-based
#-------------------------------------------------------------------------------
metabolite_MNet_venn <- all_filter_metabolite %>%
filter(software=="MNet") %>%
pull(pathway)
metabolite_multigsea_same_venn <- all_filter_metabolite %>%
filter(software == "multiGSEA-samedatabase") %>%
pull(pathway)
metabolite_multigsea_diff_venn <- all_filter_metabolite %>%
filter(software == "multiGSEA-diffdatabase") %>%
pull(pathway)
a<-venn.diagram(list(metabolite_mnet=metabolite_MNet_venn,
metabolite_multigsea_same=metabolite_multigsea_same_venn,
metabolite_multigsea_diff=metabolite_multigsea_diff_venn),
filename=NULL,fill=c("#EF2C2B","#FFC839","#4FBD81"),
alpha=0.9)
pdf("result/figure3/3b.GSEA_metabolite_venn.pdf",width=5,height = 5)
grid.draw(a)
dev.off()
#-------------------------------------------------------------------------------
# gene-based
#-------------------------------------------------------------------------------
gene_MNet_venn <- all_filter_gene %>%
filter(software=="MNet") %>%
pull(pathway)
gene_multigsea_same_venn <- all_filter_gene %>%
filter(software == "multiGSEA-samedatabase") %>%
pull(pathway)
gene_multigsea_diff_venn <- all_filter_gene %>%
filter(software == "multiGSEA-diffdatabase") %>%
pull(pathway)
a<-venn.diagram(list(gene_mnet=gene_MNet_venn,
gene_multigsea_same=gene_multigsea_same_venn,
gene_multigsea_diff=gene_multigsea_diff_venn),
filename=NULL,fill=c("#EF2C2B","#FFC839","#4FBD81"),
alpha=0.9)
pdf("result/figure3/3c.GSEA_gene_venn.pdf",width=5,height = 5)
grid.draw(a)
dev.off()
#-------------------------------------------------------------------------------
# integrated metabolite-gene analyses
#-------------------------------------------------------------------------------
extended_MNet_venn <- all_filter_extended %>%
filter(software=="MNet") %>%
pull(pathway)
extended_multigsea_same_venn <- all_filter_extended %>%
filter(software == "multiGSEA-samedatabase") %>%
pull(pathway)
extended_multigsea_diff_venn <- all_filter_extended %>%
filter(software == "multiGSEA-diffdatabase") %>%
pull(pathway)
a<-venn.diagram(list(extended_mnet=extended_MNet_venn,
extended_multigsea_same=extended_multigsea_same_venn,
extended_multigsea_diff=extended_multigsea_diff_venn),
filename=NULL,fill=c("#EF2C2B","#FFC839","#4FBD81"),
alpha=0.9)
pdf("result/figure3/3d.GSEA_extended_venn.pdf",width=5,height = 5)
grid.draw(a)
dev.off()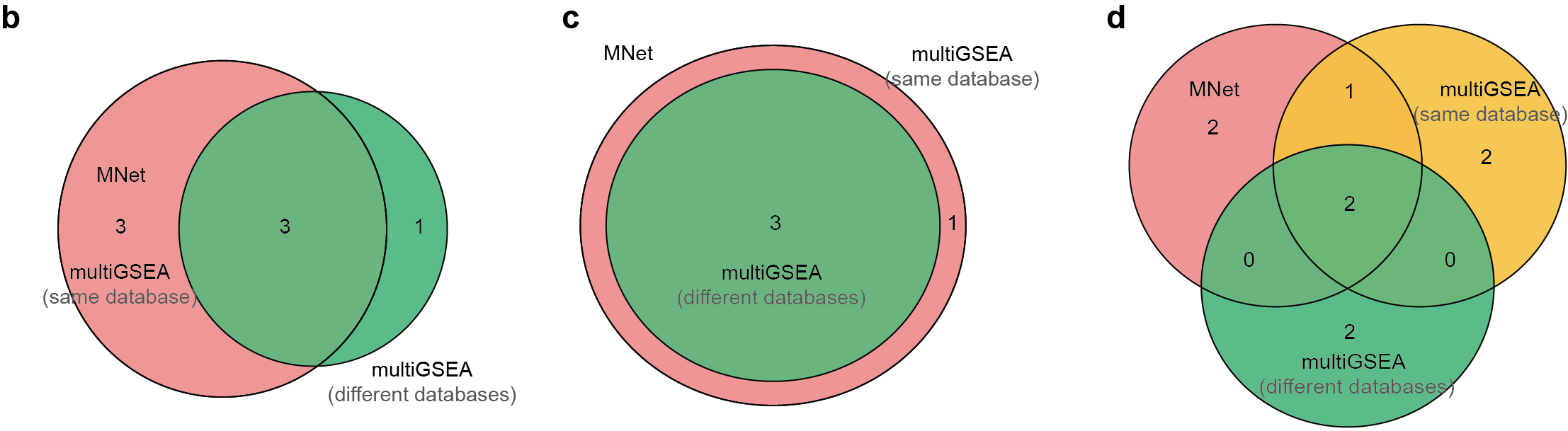
3.4 Figure 4
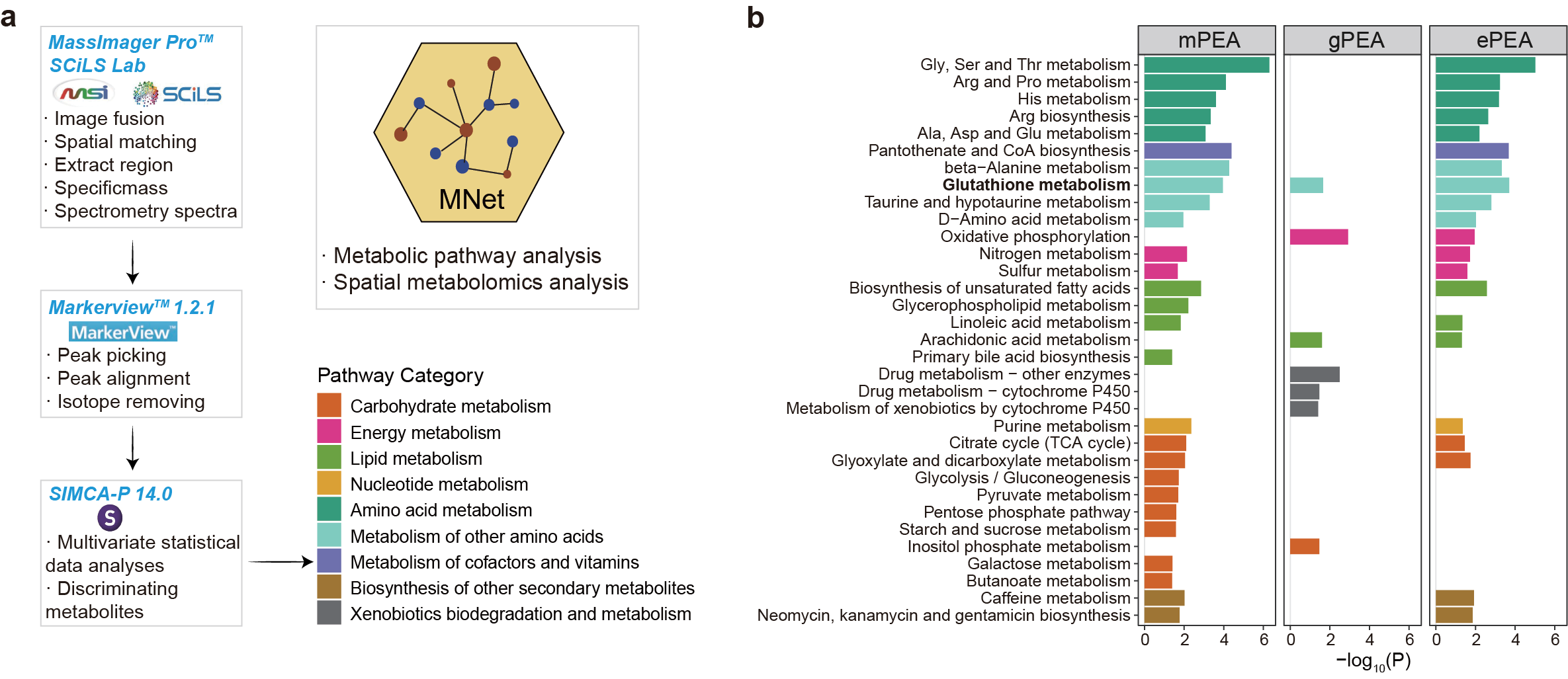
Fig. 4 | Spatial metabolomics uncovers tumour microenvironment heterogeneity.
Gastric cancer analysis workflow.
Integrated pathway enrichment (ePEA) reveals dysregulation beyond originally reported pathways, with glutathione metabolism showing strongest joint enrichment, driving intratumour heterogeneity.
3.4.1 (4b) Pathway enrichment analysis
Integrated pathway enrichment (ePEA) reveals dysregulation beyond originally reported pathways, with glutathione metabolism showing strongest joint enrichment, driving intratumour heterogeneity.
library(ggplot2)
library(dplyr)
library(MNet)
#-------------------------------------------------------------------------------
# Input metabolome and transcriptome data for pathway analysis
#-------------------------------------------------------------------------------
kegg_pathway_uniq <- PathwayExtendData %>%
dplyr::select(kegg_pathwayname,kegg_category) %>%
dplyr::rename("PATHWAY"="kegg_pathwayname") %>%
dplyr::rename("pathway_type"="kegg_category") %>%
unique()
dat_gene <- data.table::fread("input/special-gene-pathway.txt") %>%
as.data.frame() %>%
dplyr::select(name,pvalue) %>%
mutate(type="gene")
dat_metabolite <- data.table::fread("input/special-metabolite-pathway.txt") %>%
as.data.frame() %>%
dplyr::select(name,pvalue) %>%
mutate(type="metabolite")
dat_all <- data.table::fread("input/special-all-pathway.txt") %>%
as.data.frame() %>%
dplyr::select(name,pvalue) %>%
mutate(type="all")
result_1 <- rbind(dat_gene,dat_metabolite,dat_all) %>%
dplyr::left_join(kegg_pathway_uniq,by=c("name"="PATHWAY")) %>%
dplyr::arrange(pvalue)
pathway_hh <- unique(result_1$pathway_type)
result_1 <- result_1 %>%
dplyr::arrange(match(pathway_type,pathway_hh))
result_1$name <- factor(result_1$name,levels = rev(unique(result_1$name)))
result_1$pathway_type <- factor(result_1$pathway_type,levels=unique(kegg_pathway_uniq$pathway_type))
result_1$type <- factor(result_1$type,levels=c("metabolite","gene","all"))
#-------------------------------------------------------------------------------
# Barplot
#-------------------------------------------------------------------------------
colp <- c("Amino acid metabolism" ="#1B9E77",
"Carbohydrate metabolism"="#D95F02",
"Glycan biosynthesis and metabolism"="#1F78B4",
"Metabolism of cofactors and vitamins"="#7570B3",
"Metabolism of terpenoids and polyketides"="#BC80BD",
"Metabolism of other amino acids"="#8DD3C7",
"Energy metabolism"="#E7298A",
"Lipid metabolism"="#66A61E",
"Nucleotide metabolism"="#E6AB02",
"Biosynthesis of other secondary metabolites"="#A6761D",
"Xenobiotics biodegradation and metabolism"="#666666")
p1 <- ggplot(result_1,aes(name,-log10(pvalue)))+
geom_bar(stat="identity",aes(fill=pathway_type))+
scale_fill_manual(values=colp,name="Pathway Category")+
coord_flip()+
theme_bw()+
facet_wrap(vars(type),nrow=1)+
labs(x=NULL)
ggsave("result/figure4/4b.special-ePEA.pdf",p1,width=10,height = 5.5)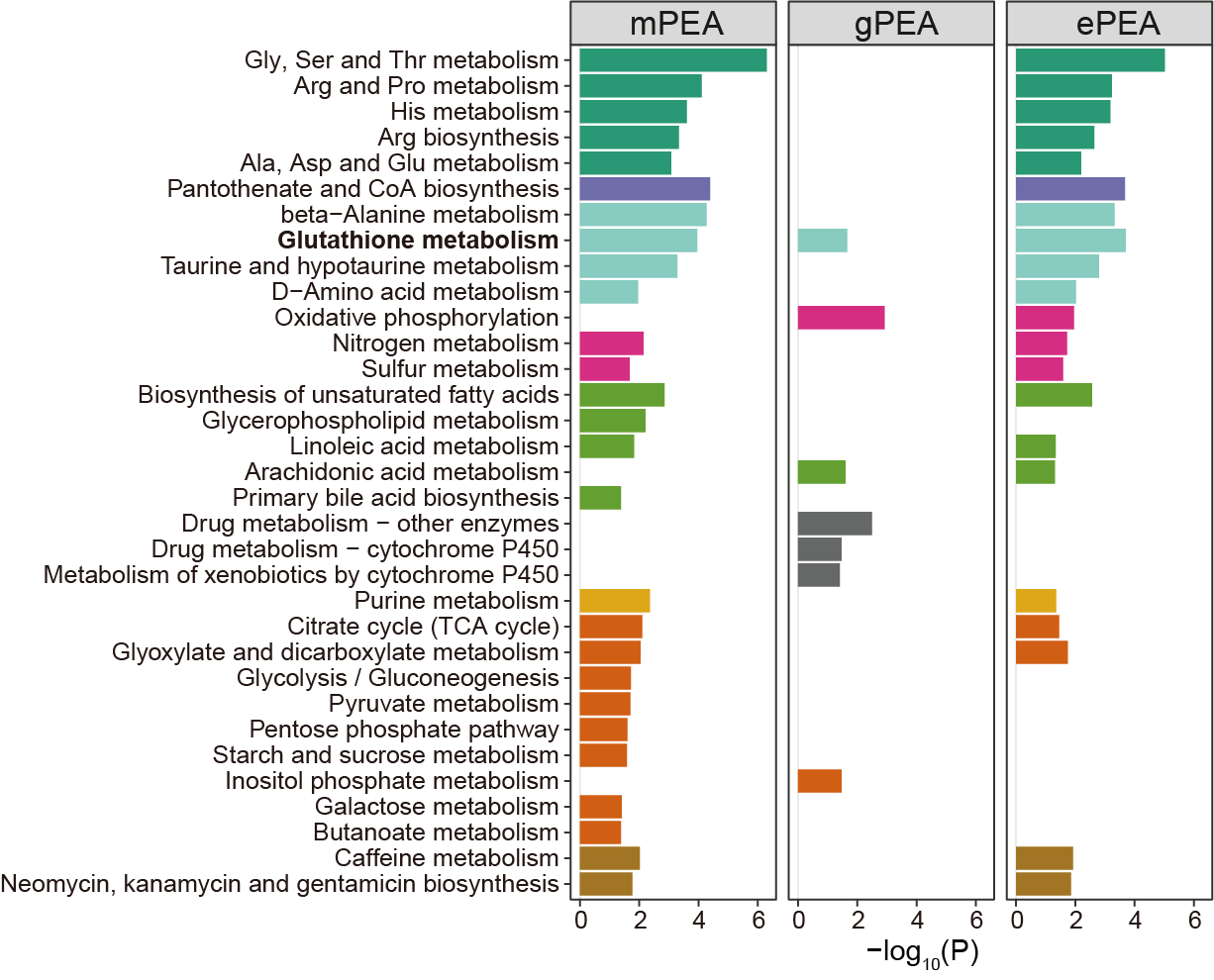
3.5 Figure 5
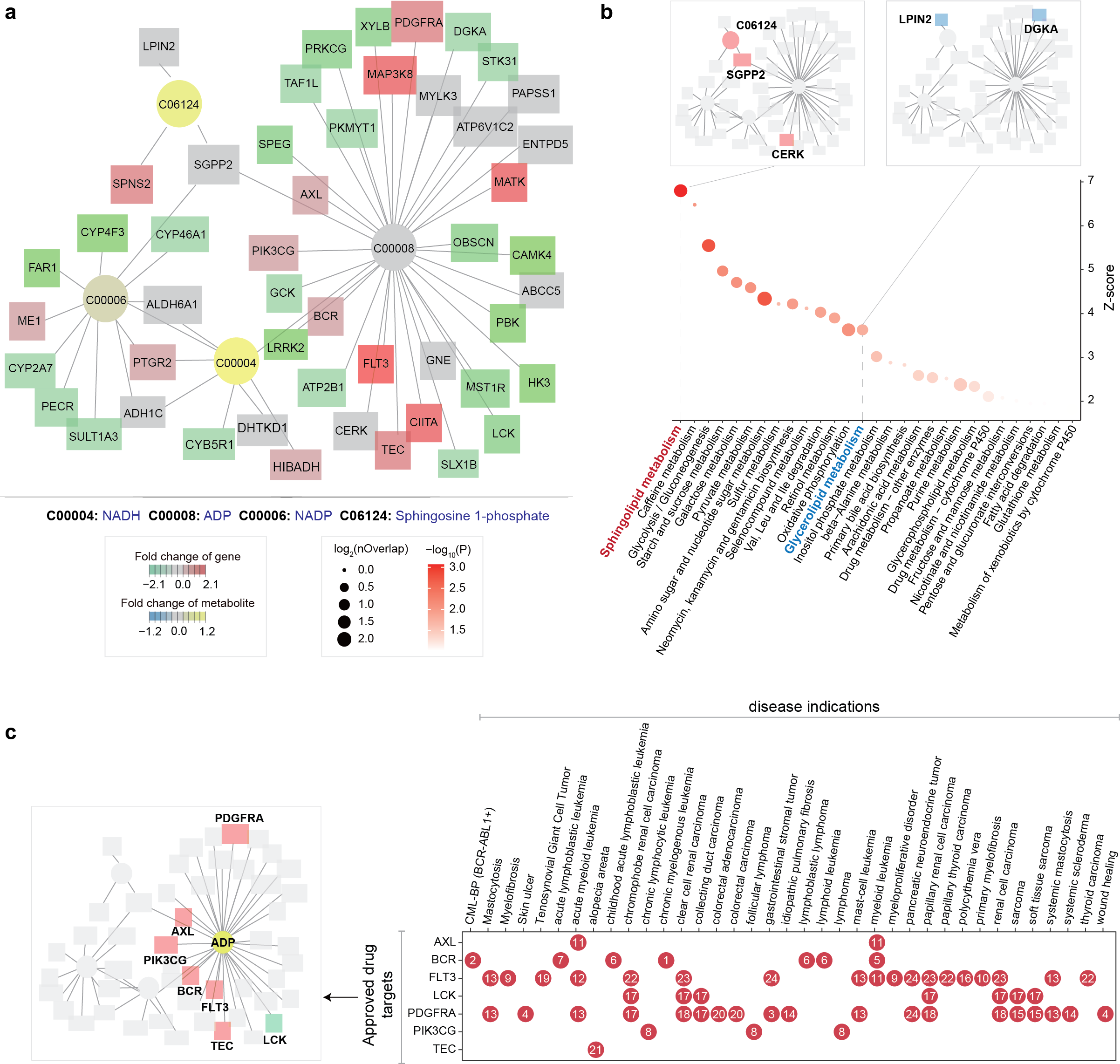
Fig. 5 | Leukaemia subnetwork analysis reveals therapeutic targets.
Core metabolic subnetwork differentiating early T-cell acute lymphoblastic leukaemia (ETP-ALL) from non-ETP-ALL. Circular nodes: metabolites (annotated with KEGG IDs); square nodes: genes. Colour gradient indicates log2(fold change) values: red (upregulated genes), green (downregulated genes), yellow (increased metabolites), and blue (decreased metabolites). List of metabolites and genes detailed in Supplementary Data 3.
Extended pathway enrichment validating sphingolipid and glycerolipid dysregulation. Inserted are the same subnetwork layout but only labelled by their respective member genes.
Therapeutic target identification via a heatmap-style plot, depicting disease indications (x-axis) against approved drug targets (y-axis; subnetwork genes are labelled on the left panel). Red indexed dots denote repurposable drug targets, with detailed information on approved drugs provided in Supplementary Data 4. Blast Phase Chronic Myelogenous Leukemia, BCR-ABL1 Positive: CML-BP (BCR-ABL1+).
3.5.1 (5a) Core metabolic subnetwork
Core metabolic subnetwork differentiating early T-cell acute lymphoblastic leukaemia (ETP-ALL) from non-ETP-ALL. Circular nodes: metabolites (annotated with KEGG IDs); square nodes: genes. Colour gradient indicates log2(fold change) values: red (upregulated genes), green (downregulated genes), yellow (increased metabolites), and blue (decreased metabolites). List of metabolites and genes detailed in Supplementary Data 3.
#-------------------------------------------------------------------------------
# Input metabolome and transcriptome data for subnetwork analysis
#-------------------------------------------------------------------------------
library(dplyr)
library(MNet)
## gene
gene_filter <- PathwayExtendData %>%
filter(type=="gene") %>%
pull(name) %>%
unique()
sample <- readxl::read_excel("input/Supplementary_Tables.xlsx",sheet=1,skip=1) %>%
as.data.frame() %>%
filter(Subtype %in% c("ETP","non_ETP")) %>%
filter(!is.na(METc_BM_D0_ID)) %>%
filter(!is.na(RNA_ID_raw)) %>%
dplyr::select(RNA_ID_raw,Subtype)
dat_gene <- readRDS("input/20231113_RNA_ALL_VST_coding.rds") %>%
as.data.frame() %>%
dplyr::select(sample$RNA_ID_raw) %>%
tibble::rownames_to_column(var="label") %>%
dplyr::filter(label %in% gene_filter) %>%
tibble::column_to_rownames("label")
group <- sample$Subtype
group[group=="ETP"] <- "tumor"
group[group=="non_ETP"] <- "normal"
result <- mlimma(dat_gene,group)
write.table(result,"result/figure5/5a.gene_ETPvsnonETP.txt",quote=F,row.names=F,sep="\t")
## metabolite
sample <- readxl::read_excel("input/Supplementary_Tables.xlsx",sheet=1,skip=1) %>%
as.data.frame() %>%
filter(Subtype %in% c("ETP","non_ETP")) %>%
filter(!is.na(METc_BM_D0_ID)) %>%
filter(!is.na(RNA_ID_raw)) %>%
dplyr::select(METc_BM_D0_ID,Subtype)
dat <- readxl::read_excel("input/Supplementary_Tables.xlsx",sheet=5,skip=1) %>%
as.data.frame() %>%
dplyr::select(Metabolite,all_of(sample$METc_BM_D0_ID)) %>%
tibble::column_to_rownames("Metabolite")
group <- sample$Subtype
group[group=="ETP"] <- "tumor"
group[group=="non_ETP"] <- "normal"
result <- mlimma(log2(dat+1),group)
write.table(result,"result/figure5/5a.metabolite_ETPvsnonETP.txt",quote=F,row.names=F,sep="\t")
## intergrated
sample <- readxl::read_excel("input/Supplementary_Tables.xlsx",sheet=1,skip=1) %>%
as.data.frame() %>%
filter(Subtype %in% c("ETP","non_ETP")) %>%
filter(!is.na(METc_BM_D0_ID)) %>%
filter(!is.na(RNA_ID_raw)) %>%
dplyr::select(RNA_ID_raw,Subtype)
dat_gene <- readRDS("input/20231113_RNA_ALL_VST_coding.rds") %>%
as.data.frame() %>%
dplyr::select(sample$RNA_ID_raw)
group <- sample$Subtype
group[group=="ETP"] <- "tumor"
group[group=="non_ETP"] <- "normal"
result <- mlimma(dat_gene,group)
write.table(result,"result/figure5/5a.gene_all_ETPvsnonETP.txt",quote=F,row.names=F,sep="\t")
#-------------------------------------------------------------------------------
# Core metabolic subnetwork analysis
#-------------------------------------------------------------------------------
library(ggplot2)
metabolite_info <- readxl::read_excel("input/Supplementary_Tables.xlsx",sheet=7,skip=1) %>%
as.data.frame() %>%
dplyr::select(Refmet_name,`KEGG ID`)
diff_gene <- data.table::fread("result/figure5/5a.gene_all_ETPvsnonETP.txt") %>%
as.data.frame()
diff_meta <- data.table::fread("result/figure5/5a.metabolite_ETPvsnonETP.txt") %>%
as.data.frame() %>%
left_join(metabolite_info,by=c("name"="Refmet_name")) %>%
filter(`KEGG ID` != "None") %>%
distinct(`KEGG ID`,.keep_all = T) %>%
dplyr::select(-name) %>%
rename("name"="KEGG ID")
names(diff_meta)[4] <- "p_value"
names(diff_gene)[4] <- "p_value"
pdf("result/T-ALL_subnetwork-50.pdf",width=10,height = 10)
a <- sNETlyser(diff_meta,diff_gene,nsize=50)
dev.off()
node <- a$node_result
write.table(node,"result/figure5/5a.T-ALL_subnetwork_node-50.txt",quote=F,row.names=F,sep="\t")
edge <- a$edge_result
write.table(edge,"result/figure5/5a.T-ALL_subnetwork_edge-50.txt",quote=F,row.names=F,sep="\t")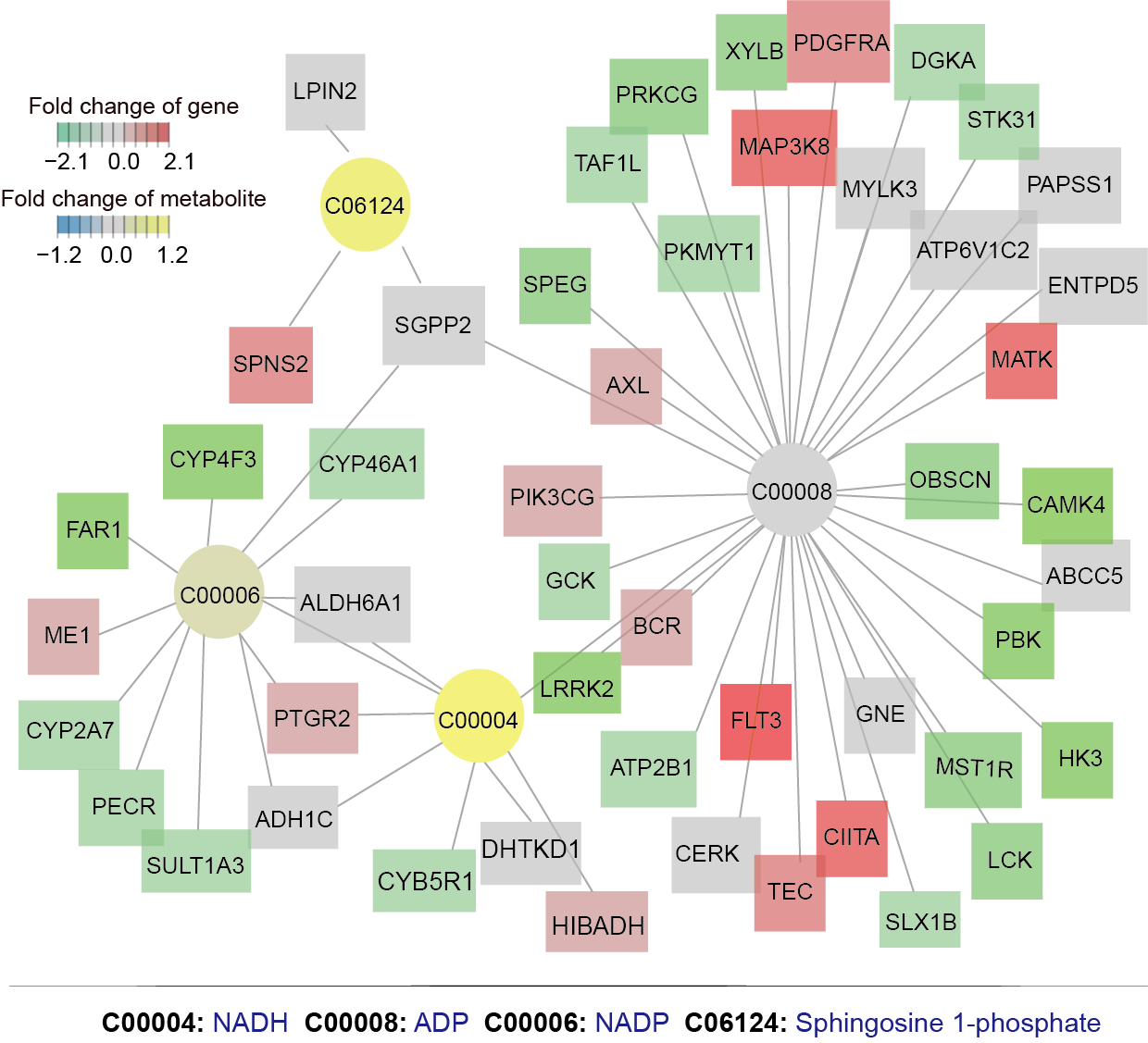
3.5.2 (5b) Extended pathway enrichment
Extended pathway enrichment validating sphingolipid and glycerolipid dysregulation. Inserted are the same subnetwork layout but only labelled by their respective member genes.
library(dplyr)
library(MNet)
library(ggplot2)
dd <- data.table::fread("result/figure5/5a.T-ALL_subnetwork_node-50.txt") %>%
as.data.frame()
## ePEA
result_ePEA <- ePEAlyser(dd$name,out = "Extended",p_cutoff = 0.1)
result_dat <- result_ePEA$output %>%
as.data.frame() %>%
dplyr::arrange(fc) %>%
dplyr::mutate(name=factor(name,levels=unique(name)))
p <- ggplot(result_dat,aes(fc,name))+
geom_point(aes(size=log2(nOverlap),color=-log10(pvalue)))+
theme_bw()+
theme(panel.border = element_blank(),panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),axis.line = element_line(colour = "black"))+
scale_color_gradient(low="white",high="red")+
labs(x=NULL,y="Pathway name")
ggsave("result/figure5/5b.T-ALL_subnetwork_ePEA-50.pdf",p,width=8,height=6)
3.6 Figure 6
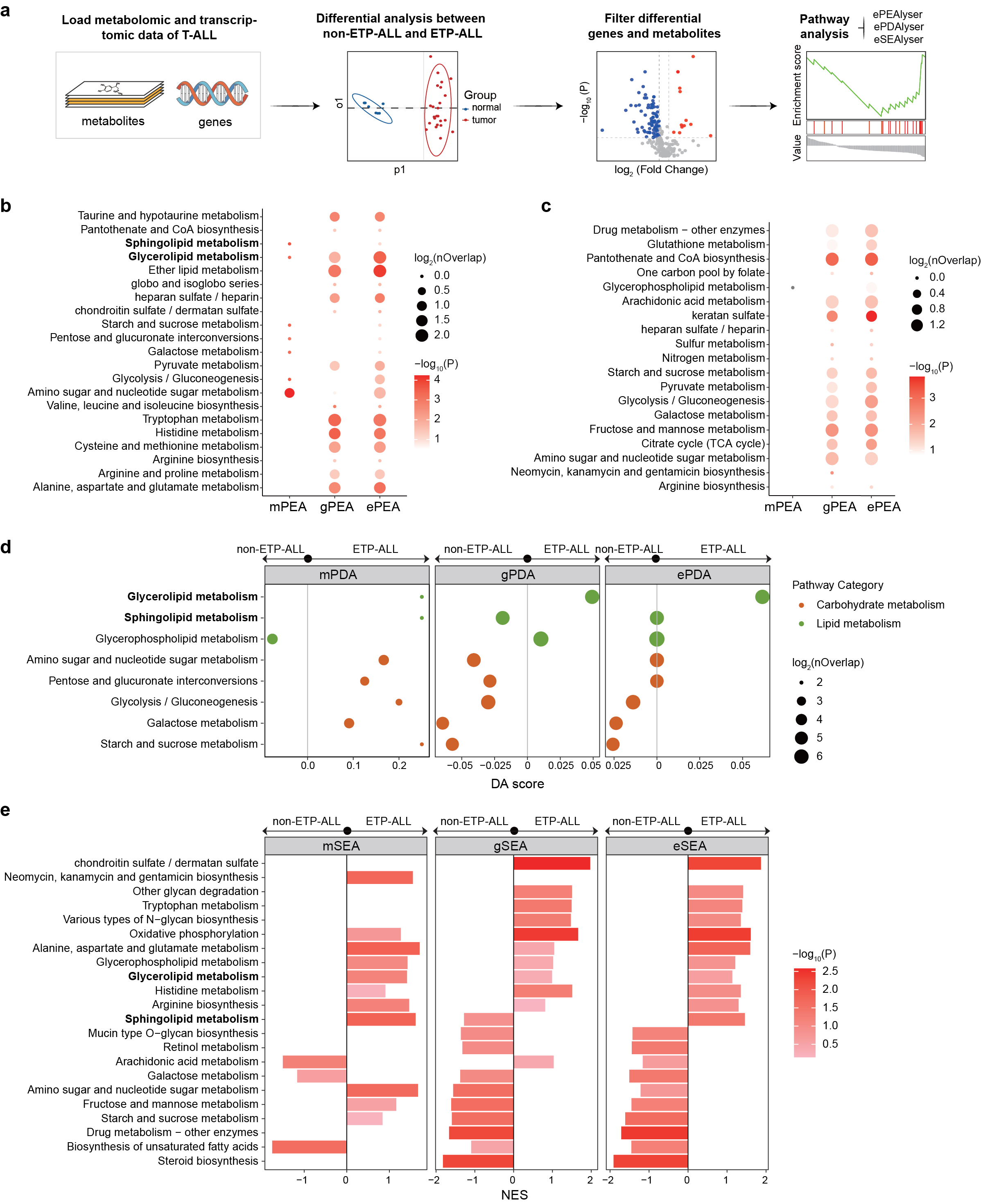
Fig. 6 | Extended pathway analyses outperform single-omics approaches in leukaemia.
- Analytical workflow.
b-c. Enrichment analysis (PEA) for (b) upregulated and (c) downregulated molecules showing ePEA’s superior sensitivity.
Differential abundance analysis (PDA) demonstrating ePDA’s increased stringency.
Set enrichment analysis (SEA) revealing eSEA’s integration capacity where mSEA/gSEA fail.
3.6.1 (6b-c) Enrichment analysis
Enrichment analysis (PEA) for (b) upregulated and (c) downregulated molecules showing ePEA’s superior sensitivity.
library(dplyr)
library(MNet)
library(ggplot2)
#-------------------------------------------------------------------------------
# PEA for upregulated and downregulated molecules
#-------------------------------------------------------------------------------
## gene
all_data <- data.table::fread("result/figure5/5a.gene_ETPvsnonETP.txt") %>%
as.data.frame()
input <- data.frame(epea_logfc = 1,epea_p = 0.05,epea_p_cutoff = 0.5)
all_data_up <- all_data %>%
filter(logFC > input$epea_logfc) %>%
filter(P.Value < input$epea_p)
result_up <- ePEAlyser(all_data_up$name,out = "gene",p_cutoff = input$epea_p_cutoff)
all_data_down <- all_data %>%
filter(logFC < -(input$epea_logfc)) %>%
filter(P.Value < input$epea_p)
result_down <- ePEAlyser(all_data_down$name,out = "gene",p_cutoff = input$epea_p_cutoff)
write.table(result_up$output,"result/figure6/6b.ePEA_gene_up.txt",quote=F,row.names=F,sep="\t")
write.table(result_down$output,"result/figure6/6c.ePEA_gene_down.txt",quote=F,row.names=F,sep="\t")
## metabolite
metabolite_info <- readxl::read_excel("input/Supplementary_Tables.xlsx",sheet=7,skip=1) %>%
as.data.frame() %>%
dplyr::select(Refmet_name,`KEGG ID`)
all_data <- data.table::fread("result/figure5/5a.metabolite_ETPvsnonETP.txt") %>%
as.data.frame() %>%
left_join(metabolite_info,by=c("name"="Refmet_name")) %>%
filter(`KEGG ID` != "None") %>%
distinct(`KEGG ID`,.keep_all = T)
input <- data.frame(epea_logfc = 0.37,epea_p = 0.05,epea_p_cutoff = 0.5)
all_data_up <- all_data %>%
filter(logFC > input$epea_logfc) %>%
filter(P.Value < input$epea_p)
result_up <- ePEAlyser(all_data_up$`KEGG ID`,out = "metabolite",p_cutoff = input$epea_p_cutoff)
all_data_down <- all_data %>%
filter(logFC < -(input$epea_logfc)) %>%
filter(P.Value < input$epea_p)
result_down <- ePEAlyser(all_data_down$`KEGG ID`,out = "metabolite",p_cutoff = input$epea_p_cutoff)
write.table(result_up$output,"result/figure6/6b.ePEA_metabolite_up.txt",quote=F,row.names=F,sep="\t")
write.table(result_down$output,"result/figure6/6c.ePEA_metabolite_down.txt",quote=F,row.names=F,sep="\t")
## integrated
metabolite_info <- readxl::read_excel("input/Supplementary_Tables.xlsx",sheet=7,skip=1) %>%
as.data.frame() %>%
dplyr::select(Refmet_name,`KEGG ID`)
all_data_metabolite <- data.table::fread("result/figure5/5a.metabolite_ETPvsnonETP.txt") %>%
as.data.frame() %>%
left_join(metabolite_info,by=c("name"="Refmet_name")) %>%
filter(`KEGG ID` != "None") %>%
distinct(`KEGG ID`,.keep_all = T) %>%
dplyr::select(-name) %>%
rename("name"="KEGG ID")
all_data_gene <- data.table::fread("result/figure5/5a.gene_ETPvsnonETP.txt") %>%
as.data.frame()
all_data <- rbind(all_data_metabolite,all_data_gene)
input <- data.frame(epda_logfc_metabolite = 0.37,epda_p_metabolite = 0.05,
epda_logfc_gene=1,epda_p_gene=0.05,epea_p_cutoff = 0.5)
all_metabolite_increase <- all_data_metabolite %>%
filter(logFC > input$epda_logfc_metabolite) %>%
filter(P.Value < input$epda_p_metabolite)
all_metabolite_decrease <- all_data_metabolite %>%
filter(logFC < -(input$epda_logfc_metabolite)) %>%
filter(P.Value < input$epda_p_metabolite)
all_gene_increase <- all_data_gene %>%
filter(logFC > input$epda_logfc_gene) %>%
filter(P.Value < input$epda_p_gene)
all_gene_decrease <- all_data_gene %>%
filter(logFC < -(input$epda_logfc_gene)) %>%
filter(P.Value < input$epda_p_gene)
all_data_up <- rbind(all_metabolite_increase,all_gene_increase)
all_data_down <- rbind(all_metabolite_decrease,all_gene_decrease)
result_up <- ePEAlyser(all_data_up$name,out = "Extended",p_cutoff = input$epea_p_cutoff)
result_down <- ePEAlyser(all_data_down$name,out = "Extended",p_cutoff = input$epea_p_cutoff)
write.table(result_up$output,"result/figure6/6b.ePEA_all_up.txt",quote=F,row.names=F,sep="\t")
write.table(result_down$output,"result/figure6/6c.ePEA_all_down.txt",quote=F,row.names=F,sep="\t")
#-------------------------------------------------------------------------------
# bubble chart for upregulated molecules
#-------------------------------------------------------------------------------
up_all <- data.table::fread("result/figure6/6b.ePEA_all_up.txt") %>%
as.data.frame() %>%
dplyr::select(name,pvalue,nOverlap) %>%
mutate(pvalue=ifelse(pvalue < 1.1e-08,1.1e-09,pvalue)) %>%
mutate(type="all")
up_gene <- data.table::fread("result/figure6/6b.ePEA_gene_up.txt") %>%
as.data.frame() %>%
dplyr::select(name,pvalue,nOverlap) %>%
mutate(type="gene")
up_metabolite <- data.table::fread("result/figure6/6b.ePEA_metabolite_up.txt") %>%
as.data.frame() %>%
dplyr::select(name,pvalue,nOverlap) %>%
mutate(type="metabolite")
up_all_filter <- up_all %>%
filter(pvalue < 0.05)
up_gene_filter <- up_gene %>%
filter(pvalue < 0.05)
up_metabolite_filter <- up_metabolite %>%
filter(pvalue < 0.05)
path_overlap <- unique(c(up_all_filter$name,up_gene_filter$name,up_metabolite_filter$name))
length(path_overlap)
result_all_filter <- up_all %>%
filter(name %in% path_overlap)
result_gene_filter <- up_gene %>%
filter(name %in% path_overlap)
result_metabolite_filter <- up_metabolite %>%
filter(name %in% path_overlap)
pathway_type <- PathwayExtendData %>%
dplyr::select(kegg_pathwayname,kegg_category) %>%
unique()
all <- rbind(result_gene_filter,result_metabolite_filter,result_all_filter) %>%
dplyr::mutate(type=factor(type,levels=c("metabolite","gene","all"))) %>%
dplyr::left_join(pathway_type,by=c("name"="kegg_pathwayname")) %>%
dplyr::arrange(kegg_category) %>%
dplyr::mutate(name=factor(name,levels=unique(name)))
p <- ggplot(all,aes(type,name))+
geom_point(aes(size=log2(nOverlap),color=-log10(pvalue)))+
theme_bw()+
theme(panel.border = element_blank(),panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),axis.line = element_line(colour = "black"))+
scale_color_gradient(low="white",high="red")+
labs(x=NULL,y="Pathway name")
ggsave("result/figure6/6b.ePEA_up_plot.pdf",p,width=8,height = 5)
#-------------------------------------------------------------------------------
# bubble plot for downregulated molecules
#-------------------------------------------------------------------------------
down_all <- data.table::fread("result/figure6/6c.ePEA_all_down.txt") %>%
as.data.frame() %>%
dplyr::select(name,pvalue,nOverlap) %>%
mutate(pvalue=ifelse(pvalue < 1.1e-08,1.1e-09,pvalue)) %>%
mutate(type="all")
down_gene <- data.table::fread("result/figure6/6c.ePEA_gene_down.txt") %>%
as.data.frame() %>%
dplyr::select(name,pvalue,nOverlap) %>%
mutate(type="gene")
down_metabolite <- data.table::fread("result/figure6/6c.ePEA_metabolite_down.txt") %>%
as.data.frame() %>%
dplyr::select(name,pvalue,nOverlap) %>%
mutate(type="metabolite")
down_all_filter <- down_all %>%
filter(pvalue < 0.05)
down_gene_filter <- down_gene %>%
filter(pvalue < 0.05)
down_metabolite_filter <- down_metabolite %>%
filter(pvalue < 0.05)
path_overlap <- unique(c(down_all_filter$name,down_gene_filter$name,down_metabolite_filter$name))
length(path_overlap)
result_all_filter <- down_all %>%
filter(name %in% path_overlap)
result_gene_filter <- down_gene %>%
filter(name %in% path_overlap)
result_metabolite_filter <- down_metabolite %>%
filter(name %in% path_overlap)
pathway_type <- PathwayExtendData %>%
dplyr::select(kegg_pathwayname,kegg_category) %>%
unique()
all <- rbind(result_gene_filter,result_metabolite_filter,result_all_filter) %>%
dplyr::mutate(type=factor(type,levels=c("metabolite","gene","all"))) %>%
dplyr::left_join(pathway_type,by=c("name"="kegg_pathwayname")) %>%
dplyr::arrange(kegg_category) %>%
dplyr::mutate(name=factor(name,levels=unique(name)))
p <- ggplot(all,aes(type,name))+
geom_point(aes(size=log2(nOverlap),color=-log10(pvalue)))+
theme_bw()+
theme(panel.border = element_blank(),panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),axis.line = element_line(colour = "black"))+
scale_color_gradient(low="white",high="red")+
labs(x=NULL,y="Pathway name")
ggsave("result/figure6/6c.ePEA_down_plot.pdf",p,width=7,height = 4.5)
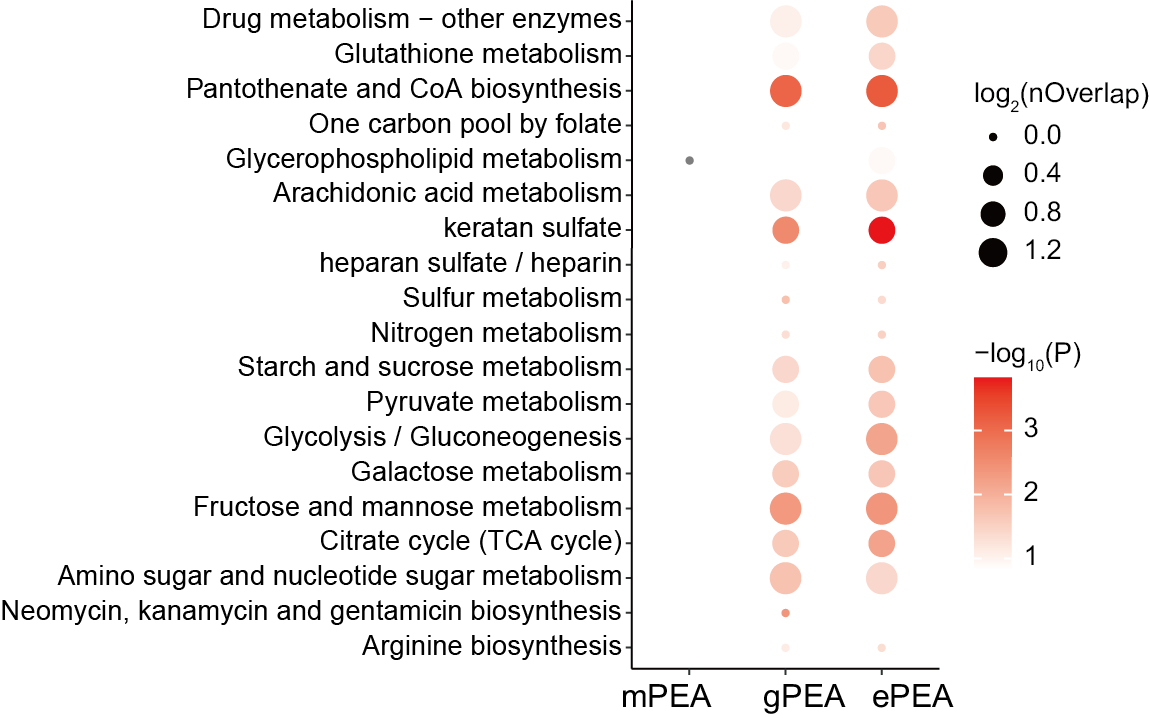
3.6.2 (6d) Differential abundance analysis
Differential abundance analysis (PDA) demonstrating ePDA’s increased stringency.
library(dplyr)
library(MNet)
library(ggplot2)
#-------------------------------------------------------------------------------
# Differential abundance analysis
#-------------------------------------------------------------------------------
## gene
diff_gene <- data.table::fread("result/figure5/5a.gene_ETPvsnonETP.txt") %>%
as.data.frame()
input <- data.frame(epda_logfc = 1,epda_padj = 0.05)
diff_gene_increase <- diff_gene %>%
filter(logFC > input$epda_logfc) %>%
filter(P.Value < input$epda_padj)
diff_gene_decrease <- diff_gene %>%
filter(logFC < -(input$epda_logfc)) %>%
filter(P.Value < input$epda_padj)
epda_res <- ePDAlyser(
c(diff_gene_increase$name),
c(diff_gene_decrease$name),
c(diff_gene$name),
sort_plot = "category",
min_measured_num = 20,
out = "gene")
write.table(epda_res$result,"result/figure6/6d.ePDA_gene.txt",quote=F,row.names = F,sep="\t")
## metabolite
metabolite_info <- readxl::read_excel("input/Supplementary_Tables.xlsx",sheet=7,skip=1) %>%
as.data.frame() %>%
dplyr::select(Refmet_name,`KEGG ID`)
all_data <- data.table::fread("result/figure5/5a.metabolite_ETPvsnonETP.txt") %>%
as.data.frame() %>%
left_join(metabolite_info,by=c("name"="Refmet_name")) %>%
filter(`KEGG ID` != "None") %>%
distinct(`KEGG ID`,.keep_all = T) %>%
dplyr::select(-name) %>%
rename("name"="KEGG ID")
input <- data.frame(epda_logfc = 0.37,epda_padj = 0.05)
all_data_increase <- all_data %>%
filter(logFC > input$epda_logfc) %>%
filter(P.Value < input$epda_padj)
all_data_decrease <- all_data %>%
filter(logFC < -(input$epda_logfc)) %>%
filter(P.Value < input$epda_padj)
epda_res <- ePDAlyser(
c(all_data_increase$name),
c(all_data_decrease$name),
c(all_data$name),
sort_plot = "category",
min_measured_num = 3,
out = "metabolite")
write.table(epda_res$result,"result/figure6/6d.ePDA_metabolite.txt",quote=F,row.names = F,sep="\t")
## integrated
all_data_metabolite <- data.table::fread("result/figure5/5a.metabolite_ETPvsnonETP.txt") %>%
as.data.frame() %>%
left_join(metabolite_info,by=c("name"="Refmet_name")) %>%
filter(`KEGG ID` != "None") %>%
distinct(`KEGG ID`,.keep_all = T) %>%
dplyr::select(-name) %>%
rename("name"="KEGG ID")
all_data_gene <- data.table::fread("result/figure5/5a.gene_ETPvsnonETP.txt") %>%
as.data.frame()
all_data <- rbind(all_data_metabolite,all_data_gene)
input <- data.frame(epda_logfc_metabolite = 0.37,
epda_p_metabolite = 0.05,
epda_logfc_gene=1,epda_p_gene=0.05)
all_metabolite_increase <- all_data_metabolite %>%
filter(logFC > input$epda_logfc_metabolite) %>%
filter(P.Value < input$epda_p_metabolite)
all_metabolite_decrease <- all_data_metabolite %>%
filter(logFC < -(input$epda_logfc_metabolite)) %>%
filter(P.Value < input$epda_p_metabolite)
all_gene_increase <- all_data_gene %>%
filter(logFC > input$epda_logfc_gene) %>%
filter(P.Value < input$epda_p_gene)
all_gene_decrease <- all_data_gene %>%
filter(logFC < -(input$epda_logfc_gene)) %>%
filter(P.Value < input$epda_p_gene)
all_data_increase <- rbind(all_metabolite_increase,all_gene_increase)
all_data_decrease <- rbind(all_metabolite_decrease,all_gene_decrease)
epda_res <- ePDAlyser(
c(all_data_increase$name),
c(all_data_decrease$name),
c(all_data$name),
sort_plot = "category",
min_measured_num = 20,
out = "Extended")
write.table(epda_res$result,"result/figure6/6d.ePDA_all.txt",quote=F,row.names = F,sep="\t")
#-------------------------------------------------------------------------------
# bubble plot for PDA
#-------------------------------------------------------------------------------
ePDA_all <- data.table::fread("result/figure6/6d.ePDA_all.txt") %>%
as.data.frame() %>%
mutate(type="all")
ePDA_gene <- data.table::fread("result/figure6/6d.ePDA_gene.txt") %>%
as.data.frame() %>%
mutate(type="gene")
ePDA_metabolite <- data.table::fread("result/figure6/6d.ePDA_metabolite.txt") %>%
as.data.frame() %>%
mutate(type="metabolite")
pathway_overlap <- intersect(intersect(ePDA_all$Pathway,ePDA_gene$Pathway),ePDA_metabolite$Pathway)
result_filter <- rbind(ePDA_all,ePDA_gene,ePDA_metabolite) %>%
filter(Pathway %in% pathway_overlap) %>%
mutate(type=factor(type,levels=c("metabolite","gene","all"))) %>%
mutate(Pathway=factor(Pathway,levels=unique(Pathway)))
colp <- c("Amino acid metabolism" ="#1B9E77",
"Carbohydrate metabolism"="#D95F02",
"Glycan biosynthesis and metabolism"="#1F78B4",
"Metabolism of cofactors and vitamins"="#7570B3",
"Metabolism of terpenoids and polyketides"="#BC80BD",
"Metabolism of other amino acids"="#8DD3C7",
"Energy metabolism"="#E7298A",
"Lipid metabolism"="#66A61E",
"Nucleotide metabolism"="#E6AB02",
"Biosynthesis of other secondary metabolites"="#A6761D",
"Xenobiotics biodegradation and metabolism"="#666666")
p <- ggplot(result_filter,aes(Pathway,DA_score,color=`Pathway Category`))+
geom_point(aes(size=log2(Measured_members_num)))+
theme_bw()+
geom_hline(yintercept=c(0),color="gray")+
theme(panel.grid.major = element_blank(),
panel.grid.minor = element_blank())+
scale_color_manual(values = colp,name = "Pathway Category")+
coord_flip()+
facet_grid(. ~ type,scales="free_x")+
labs(x="Pathway name")
ggsave("result/figure6/6d.ePDA_plot.pdf",p,width=12,height = 3)
3.6.3 (6e) Set enrichment analysis
Set enrichment analysis (SEA) revealing eSEA’s integration capacity where mSEA/gSEA fail.
library(dplyr)
library(ggplot2)
library(MNet)
#-------------------------------------------------------------------------------
# Differential abundance analysis
#-------------------------------------------------------------------------------
## gene
diff_gene <- data.table::fread("result/figure5/5a.gene_ETPvsnonETP.txt") %>%
as.data.frame() %>%
arrange(desc(logFC))
gene.data <- diff_gene$logFC
names(gene.data) <- diff_gene$name
data <- c(gene.data)
result <- eSEAlyser(data, out = "gene",minSize = 15)
write.table(result,"result/figure6/6e.eSEA_gene.txt",quote=F,row.names=F,sep="\t")
## metabolite
metabolite_info <- readxl::read_excel("input/Supplementary_Tables.xlsx",sheet=7,skip=1) %>%
as.data.frame() %>%
dplyr::select(Refmet_name,`KEGG ID`)
all_data <- data.table::fread("result/figure5/5a.metabolite_ETPvsnonETP.txt") %>%
as.data.frame() %>%
left_join(metabolite_info,by=c("name"="Refmet_name")) %>%
filter(`KEGG ID` != "None") %>%
distinct(`KEGG ID`,.keep_all = T) %>%
dplyr::select(-name) %>%
rename("name"="KEGG ID") %>%
arrange(desc(logFC))
metabolite.data <- all_data$logFC
names(metabolite.data) <- all_data$name
data <- c(metabolite.data)
result <- eSEAlyser(data, out = "metabolite",minSize = 3)
write.table(result,"result/figure6/6e.eSEA_metabolite.txt",quote=F,row.names=F,sep="\t")
## integrated
all_metabolite <- data.table::fread("result/figure5/5a.metabolite_ETPvsnonETP.txt") %>%
as.data.frame() %>%
left_join(metabolite_info,by=c("name"="Refmet_name")) %>%
filter(`KEGG ID` != "None") %>%
distinct(`KEGG ID`,.keep_all = T) %>%
dplyr::select(-name) %>%
rename("name"="KEGG ID") %>%
arrange(desc(logFC))
diff_gene <- data.table::fread("result/figure5/5a.gene_ETPvsnonETP.txt") %>%
as.data.frame() %>%
arrange(desc(logFC))
all_data <- rbind(all_metabolite,diff_gene) %>%
arrange(desc(logFC))
all.data <- all_data$logFC
names(all.data) <- all_data$name
data <- c(all.data)
result <- eSEAlyser(data, out = "Extended",minSize = 13)
write.table(result,"result/figure6/6e.eSEA_all.txt",quote=F,row.names=F,sep="\t")
#-------------------------------------------------------------------------------
# barplot for SEA
#-------------------------------------------------------------------------------
eSEA_all <- data.table::fread("result/figure6/6e.eSEA_all.txt") %>%
as.data.frame() %>%
mutate(type="all")
eSEA_gene <- data.table::fread("result/figure6/6e.eSEA_gene.txt") %>%
as.data.frame() %>%
mutate(type="gene")
eSEA_metabolite <- data.table::fread("result/figure6/6e.eSEA_metabolite.txt") %>%
as.data.frame() %>%
mutate(type="metabolite")
pathway_overlap <- rbind(eSEA_all,eSEA_gene,eSEA_metabolite) %>%
filter(pval < 0.1) %>%
pull(pathway) %>% unique()
result_filter <- rbind(eSEA_all,eSEA_gene,eSEA_metabolite) %>%
filter(pathway %in% pathway_overlap) %>%
arrange(NES) %>%
mutate(type=factor(type,levels=c("metabolite","gene","all"))) %>%
mutate(pathway=factor(pathway,levels=unique(pathway)))
p <- ggplot(result_filter,aes(pathway,NES,fill=-log10(pval)))+
geom_bar(stat="identity")+
theme_bw()+
geom_hline(yintercept=c(0))+
theme(panel.grid.major = element_blank(),
panel.grid.minor = element_blank())+
scale_fill_gradient(low="pink",high="red")+
coord_flip()+
facet_grid(. ~ type,scales="free_x")+
labs(x="Pathway name")
ggsave("result/figure6/6e.eSEA_plot.pdf",p,width=10,height = 5)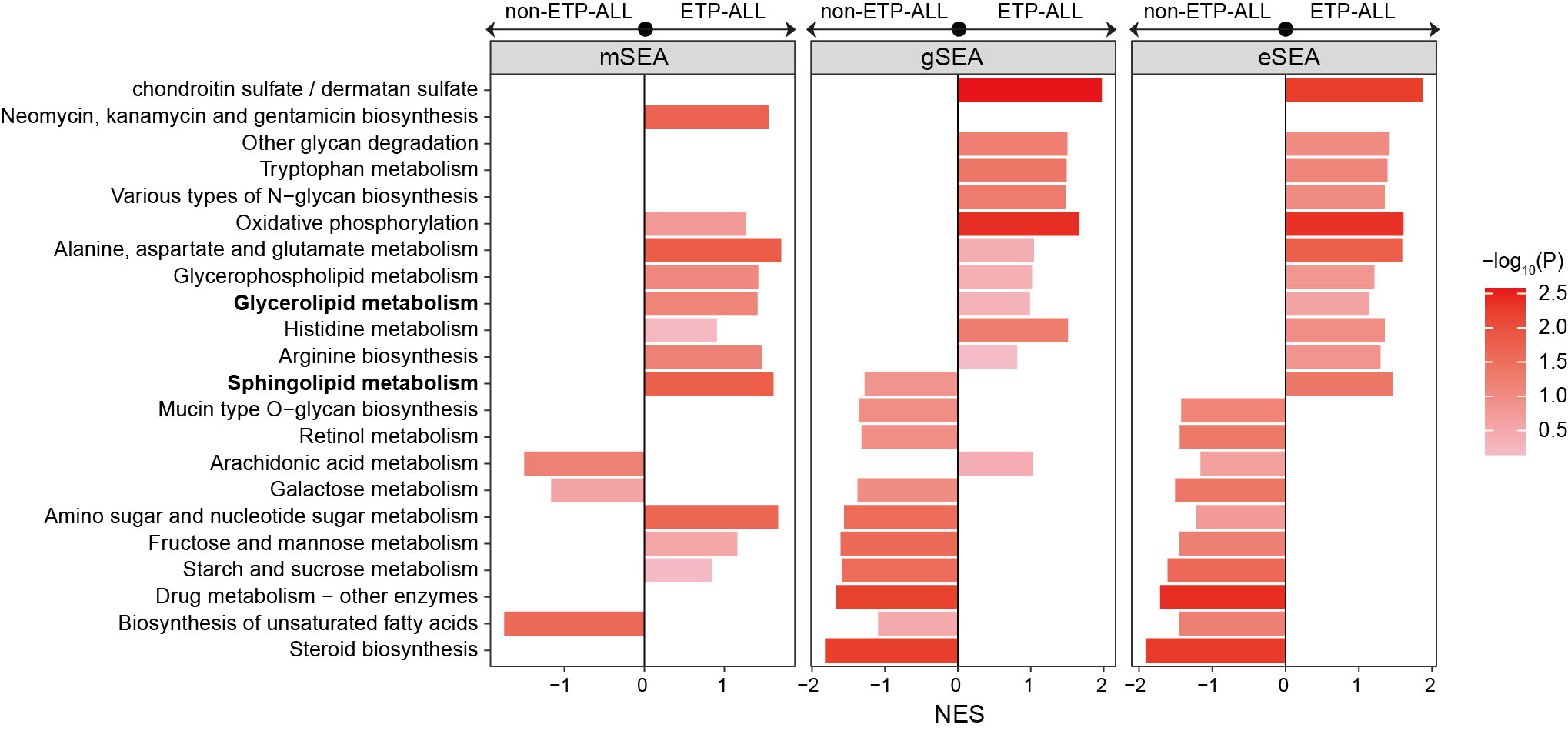
3.7 Need help?
If you have any questions about MNet, please don’t hesitate to email me (guituant2009@163.com).